- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Kinetic Lactate Dehydrogenase Assay for Detection of Cell Damage in Primary Neuronal Cell Cultures
Published: Vol 7, Iss 11, Jun 5, 2017 DOI: 10.21769/BioProtoc.2308 Views: 11850
Reviewed by: Soyun KimAnna La TorreAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The aim of many in vitro models of acute or chronic degenerative disorders in the neurobiology field is the assessment of survival or damage of neuronal cells. Damage of cells is associated with loss of outer cell membrane integrity and leakage of cytoplasmic cellular proteins. Therefore, activity assays of cytoplasmic enzymes in supernatants of cell cultures serve as a practicable tool for quantification of cellular injury (Koh and Choi, 1987; Bruer et al., 1997). Lactate dehydrogenase (LDH) is such a ubiquitously expressed cytosolic enzyme, which is very stable due to a very long protein half-life (Hsieh and Blumenthal, 1956; Koh and Cotman, 1992; Koh et al., 1995).
Keywords: LDH assayBackground
LDH catalyzes the formation of lactate and Nicotinamidadenindinucleotid (NAD+) from pyruvate and reduced Nicotinamidadenindinucleotid (NADH) in a reversible biochemical reaction. NADH has an absorption on the wavelength of 340 nm. The basis of this kinetic LDH activity assay is the decrease of optical density at the specific wave length caused by a decrease of NADH. The amount of LDH in the supernatant is calculated using a standard enzyme solution with known LDH activity. Different cell densities or metabolic activation rates might be confounders; therefore normalization of LDH activity is recommended. This is achieved by assessment of LDH activity after outer cell membrane lysis that does not block LDH activity itself (‘full kill’ with 0.5% Triton-X). Finally, percentage of absolute LDH activity from LDH activity by full kill indicates the rate of damaged or dead cells in the cell culture well.
Materials and Reagents
- 96 well plate, flat bottom (SARSTEDT, catalog number: 82.1581 )
- Pipette tips
- Neuronal cell cultures which was treated by noxes (e.g., oxygen and glucose deprivation, glutamate or other toxins) (Koh and Choi, 1987; Bruer et al., 1997; Harms et al., 2001; Ruscher et al., 2002; Harms et al., 2004; Meisel et al., 2006; Harms et al., 2007; Datwyler et al., 2011; Schweizer et al., 2015; Donath et al., 2016)
Note: This protocol works for primary neuronal cultures that were derived from various regions of the brain including septum, hippocampus, striatum, spinal cord, cortex, cerebellum or raphe nuclei and that were seeded in various densities or with various media formulations or coatings of the plates. However, we achieve best results with cultures that are at least cultivated for more than 7 days in vitro (DIV) and that were seeded in a reasonable density as illustrated below (Figures 1 and 2). These cultures contain approximately 10% glial cells and this does not impact on the possibility to analyze LDH release in the supernatant medium.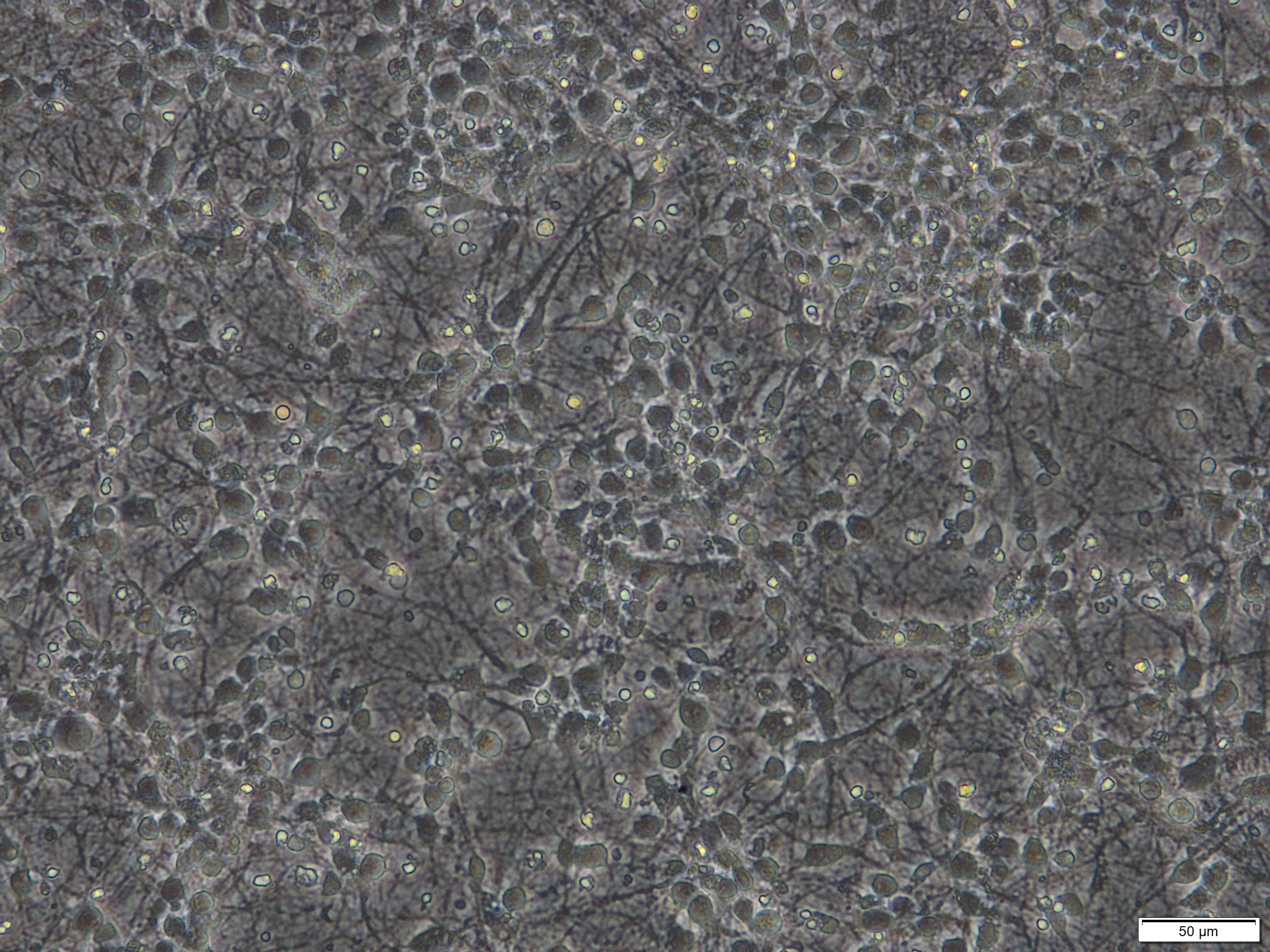
Figure 1. Primary cortical neuronal cultures derived from mouse E15 embryos after 7 days in vitro (DIV). Scale bar = 50 µm.
Figure 2. Primary cortical neuronal cultures that were treated with 50 µM glutamate for 24 h on DIV 8. Scale bar = 50 µm. - Triton X-100 solution (Sigma-Aldrich, catalog number: 93443 )
- Potassium phosphate monobasic (KH2PO4) (MW 136.1) (Sigma-Aldrich, catalog number: P5655 )
- Dibasic potassium phosphate (K2HPO4; MW 174.2) or potassium phosphate trihydrate (K2HPO4·3H2O; MW 228.2) (Sigma-Aldrich catalog number: P5504 )
- Distilled water
- Na-pyruvate (MW 110) (Sigma-Aldrich, catalog number: P2256 )
- LDH standard (TruCal U) (DiaDys Diagnostic Systems, catalog number: 5 9100 99 10 063 )
- -NADH (MW 709.4, reduced form) (Sigma-Aldrich, catalog number: N8129 )
- 10x LDH-buffer (see Recipes)
- 1x LDH-buffer (see Recipes)
- LDH-standard (see Recipes)
- Na-pyruvate-solution (see Recipes)
- -NADH-solution (see Recipes)
Equipment
- Pipette
- CO2-incubator for cell cultures
- Multi-pipette (Eppendorf, model: Multipette® plus )
- Spectrophotometer for 96 well plate that can measure 340 nm and kinetic measurement (e.g., Dynex Technologies, model: MRX Microplate Reader )
Procedure
- Preparation of solutions (see Recipes)
- Measurement
- Warm up NADH and an appropriate aliquot of Na-pyruvate-solution (for one 96 well plate you need about 2.5 ml Na-pyruvate-solution) to room temperature or max. 37 °C, thaw appropriate amount of LDH-standard aliquots.
- Pipette 50 µl cell culture medium (probe) in each well. A duplicate measurement of each cell culture well is recommended.
- Pipette 25 µl LDH standard in 2 wells per plate. It is very important that you perform a standard measurement on each 96 well plate.
- Add 200 µl NADH solution to each well and remove bubbles if necessary. The final concentration of NADH in the assay is 153.8 µM.
- Add 25 µl Na-pyruvate solution to each well. The enzymatic reaction starts promptly. The final concentration of Na-pyruvate in the assay is 2.063 mM. Do not mix samples with pipets. Mixing should be initiated by using automated shaking by the plate reader before and in between of each measurement cycle. The reaction is within the linear range, if excess NADH is available for the enzymatic reaction. This lasts about 7-10 min. Ideally, pipetting of pyruvate and measurement of all cycles should be accomplished within this period. Do not start the reaction in more than one plate at once. It might be necessary to dilute the sample as specified in Figure 3.
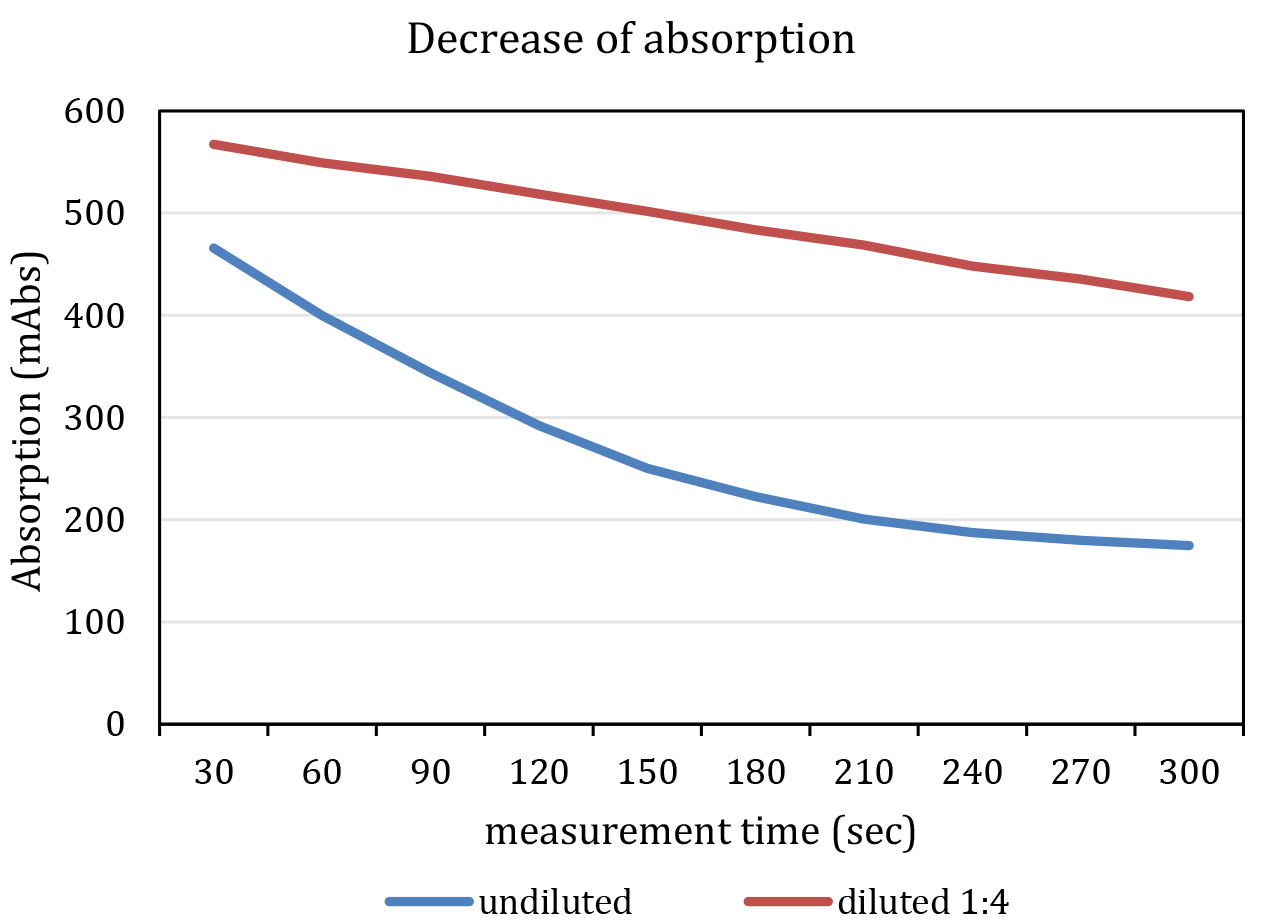
Figure 3. Representative graph of a measurement. The blue line indicates an undiluted and the red line a diluted (F1:4, red) kinematic curve. Note that the undiluted sample shows a non-linear decrease in absorption at 340 nm. - Immediately start measurement of absorption with following parameters:
- Wavelength: 340 nm.
- Measurement cycles: 10.
- Cycle time: 30 sec.
- Shaking time: 5 sec.
- Temperature: 37 °C (see also Notes).
- Total lysis
- Add Triton X-100 to the rest of cell culture medium. The final concentration should be 0.5%.
- Incubate on 37 °C for 20 min. It is possible to incubate a longer time until to 4-6 h or overnight. This is not critical as long as you treat all groups equally.
- Repeat steps 2b-2f from paragraph 2 (Measurement).
Data analysis
- Calculate the decrease of absorption for each well in mAbs/min. In our hands the value is about 10-50 mAbs/min.
Note: Check, if the decrease of absorption is linear over the whole time. Otherwise, reduce the volume of probe or dilute them. - The decrease of absorption in LDH standard equates an LDH activity about 500 U/l. The LDH activity in standard is a little bit different in different batches. Calculate according to manufactures specifications.
- Calculate the LDH activity for your probes using a rule of proportion.
Example:- Decrease of absorption in probe: 15 mAbs/min.
- Decrease of absorption in LDH standard: 43 mAbs/min.
- LDH activity in standard 466 U/l.
- Decrease of absorption in probe: 15 mAbs/min.
- Adjust the LDH activity for probe on the relation of probe volume and LDH standard volume.
- Finally, calculate LDH activity as a percentage of LDH activity in the same cell culture sample after assessing LDH activity after total LDH release (‘full kill’).
Notes
- The reaction rate is dependent on the temperature. In case of high LDH activity in your probe and an additional high temperature, an exhaustion of -NADH is possible and leads to a non-linear decrease of absorption.
- Assessment of total LDH release after cell lysis (‘full kill’) is recommended for each cell culture well. There are some treatments, which lead to an increase of protein synthesis in cells, so that the amount of total LDH in the cells is increased as well. Normalization helps for better correlation as an indirect measure of cell death in such cases.
- It is possible to store samples of supernatants at 4 °C if samples are sealed to avoid evaporation for two to three days with a minimal loss of LDH activity. Do not freeze samples.
Recipes
- 10x LDH-buffer
45.3 g KH2PO4
116.1 g K2HPO4 or 152 g K2HPO4·3H2O
Dissolve in about 800 ml distilled water, adjust pH to 7.4 and fill up to 1,000 ml with distilled water
Store at 4 °C, The solution is stable for 12 month - 1x LDH-buffer
Dilute 10x LDH buffer with distilled water 1:10
Store at 4 °C, long-term storage up to 12 months is possible - LDH-standard
Dissolve lysate of enzyme according to manufacture's protocol, prepare aliquots of 105 µl (that’s for measurement of two plates) and store at -20 °C. Avoid freeze and thaw cycles - Na-pyruvate-solution
Dissolve 1.25 g Na-pyruvate in 500 ml 1x LDH-buffer, results in 22.7 mM Na-pyruvate-solution
Store at 4 °C. The solution is stable for 18 month - -NADH-solution
Dissolve 3 mg -NADH in 20 ml 1x LDH-buffer, results in 211.4 µM solution
Note: 20 ml is the amount for one 96 well plate. This solution is stable only for 2 days at 4 °C.
Acknowledgments
This work was supported by the German Research Foundation (SFB TRR43 and HA5741/1-2 to Christoph Harms), the Federal Ministry of Education and Research (01 EO 08 01) for funding of the Center for Stroke Research Berlin (project ‘SUMO and stroke’ to Christoph Harms), and Berlin Institute of Health, TRG7, TP1 to Christoph Harms.
References
- Bruer, U., Weih, M. K., Isaev, N. K., Meisel, A., Ruscher, K., Bergk, A., Trendelenburg, G., Wiegand, F., Victorov, I. V. and Dirnagl, U. (1997). Induction of tolerance in rat cortical neurons: hypoxic preconditioning. FEBS Lett 414(1): 117-121.
- Datwyler, A. L., Lattig-Tunnemann, G., Yang, W., Paschen, W., Lee, S. L., Dirnagl, U., Endres, M. and Harms, C. (2011). SUMO2/3 conjugation is an endogenous neuroprotective mechanism. J Cereb Blood Flow Metab 31(11): 2152-2159.
- Donath, S., An, J., Lee, S. L., Gertz, K., Datwyler, A. L., Harms, U., Muller, S., Farr, T. D., Fuchtemeier, M., Lattig-Tunnemann, G., Lips, J., Foddis, M., Mosch, L., Bernard, R., Grittner, U., Balkaya, M., Kronenberg, G., Dirnagl, U., Endres, M. and Harms, C. (2016). Interaction of ARC and Daxx: A novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. J Neurosci 36(31): 8132-8148.
- Harms, C., Albrecht, K., Harms, U., Seidel, K., Hauck, L., Baldinger, T., Hubner, D., Kronenberg, G., An, J., Ruscher, K., Meisel, A., Dirnagl, U., von Harsdorf, R., Endres, M. and Hortnagl, H. (2007). Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J Neurosci 27(17): 4562-71.
- Harms, C., Bosel, J., Lautenschlager, M., Harms, U., Braun, J. S., Hortnagl, H., Dirnagl, U., Kwiatkowski, D. J., Fink, K. and Endres, M. (2004). Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Mol Cell Neurosci 25(1): 69-82.
- Harms, C., Lautenschlager, M., Bergk, A., Katchanov, J., Freyer, D., Kapinya, K., Herwig, U., Megow, D., Dirnagl, U., Weber, J. R. and Hortnagl, H. (2001). Differential mechanisms of neuroprotection by 17 β-estradiol in apoptotic versus necrotic neurodegeneration. J Neurosci 21(8): 2600-2609.
- Hsieh, K. M. and Blumenthal, H. T. (1956). Serum lactic dehydrogenase levels in various disease states. Proc Soc Exp Biol Med 91(4): 626-630.
- Koh, J. Y. and Choi, D. W. (1987). Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods 20(1): 83-90.
- Koh, J. Y. and Cotman, C. W. (1992). Programmed cell death: its possible contribution to neurotoxicity mediated by calcium channel antagonists. Brain Res 587(2): 233-240.
- Koh, J. Y., Gwag, B. J., Lobner, D. and Choi, D. W. (1995). Potentiated necrosis of cultured cortical neurons by neurotrophins. Science 268(5210): 573-575.
- Meisel, A., Harms, C., Yildirim, F., Bosel, J., Kronenberg, G., Harms, U., Fink, K. B. and Endres, M. (2006). Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem 98(4): 1019-1031.
- Ruscher, K., Freyer, D., Karsch, M., Isaev, N., Megow, D., Sawitzki, B., Priller, J., Dirnagl, U. and Meisel, A. (2002). Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci 22(23): 10291-301.
- Schweizer, S., Harms, C., Lerch, H., Flynn, J., Hecht, J., Yildirim, F., Meisel, A. and Marschenz, S. (2015). Inhibition of histone methyltransferases SUV39H1 and G9a leads to neuroprotection in an in vitro model of cerebral ischemia. J Cereb Blood Flow Metab 35(10): 1640-1647.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Freyer, D. and Harms, C. (2017). Kinetic Lactate Dehydrogenase Assay for Detection of Cell Damage in Primary Neuronal Cell Cultures. Bio-protocol 7(11): e2308. DOI: 10.21769/BioProtoc.2308.
- Donath, S., An, J., Lee, S. L., Gertz, K., Datwyler, A. L., Harms, U., Muller, S., Farr, T. D., Fuchtemeier, M., Lattig-Tunnemann, G., Lips, J., Foddis, M., Mosch, L., Bernard, R., Grittner, U., Balkaya, M., Kronenberg, G., Dirnagl, U., Endres, M. and Harms, C. (2016). Interaction of ARC and Daxx: A novel endogenous target to preserve motor function and cell loss after focal brain ischemia in mice. J Neurosci 36(31): 8132-8148.
Category
Neuroscience > Nervous system disorders > Cellular mechanisms
Biochemistry > Protein > Quantification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.