- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Detection of Neutrophil Traps and Post-attack Cell Wall Changes in Candida Hyphae

Published: Vol 7, Iss 7, Apr 5, 2017 DOI: 10.21769/BioProtoc.2213 Views: 10903
Reviewed by: Alka MehraSaskia F. ErttmannSadri Znaidi
Original research article
The authors used this protocol in:

May 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
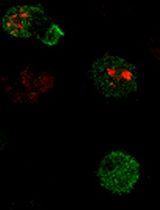
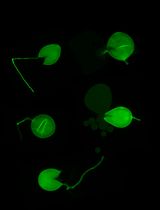

Abstract
In this protocol we describe how to visualize neutrophil extracellular traps (NETs) and fungal cell wall changes in the context of the coculture of mouse neutrophils with fungal hyphae of Candida albicans. These protocols are easily adjusted to test a wide array of hypotheses related to the impact of immune cells on fungi and the cell wall, making them promising tools for exploring host-pathogen interactions during fungal infection.
Keywords: FungiBackground
C. albicans is a polymorphic opportunistic yeast and neutrophils are immune cells critical for defense against this and other fungal pathogens (Brown et al., 2012; Lionakis and Netea, 2013). NETs are a potential defense mechanism that can be deployed against pathogens and it has been suggested that they are preferentially deployed against microbial cells such as C. albicans hyphae that are too large to phagocytose (Urban et al., 2006; Bruns et al., 2010; Branzk et al., 2014; Rohm et al., 2014). NETs have been shown to contain a number of components including myeloperoxidase, extracellular DNA and citrullinated histones (Amulic et al., 2012; Branzk and Papayannopoulos, 2013). For positive identification of NETs, the standard in both in vitro and in vivo experiments includes staining for and demonstrating the colocalization of these markers. While the exact contribution of NETs to defense against C. albicans infection is not well understood, our group has demonstrated they can provoke stress responses and cell wall rearrangement in C. albicans hyphae. Specifically, NET attack results in greater chitin deposition and β-glucan exposure as shown schematically in Figure 1. These polysaccharides normally lie underneath the mannan layer, and their exposure can change immune recognition (Perez-Garcia et al., 2011). The basic assays described here were used extensively to probe this subject by our group (Hopke et al., 2016). While outlined here for the purpose of detecting NETs and fungal cell wall changes, this protocol is easily tweaked to leverage many combinations of chemical inhibitors, transgenic or knockout fungal strains or mouse neutrophils and other staining targets to test a wide array of hypotheses (Hopke et al., 2016). This protocol therefore represents a promising method to further elucidate the impact immune cells have on the C. albicans cell wall, its stress response and the importance of altered epitope exposure to host defense against fungal infection.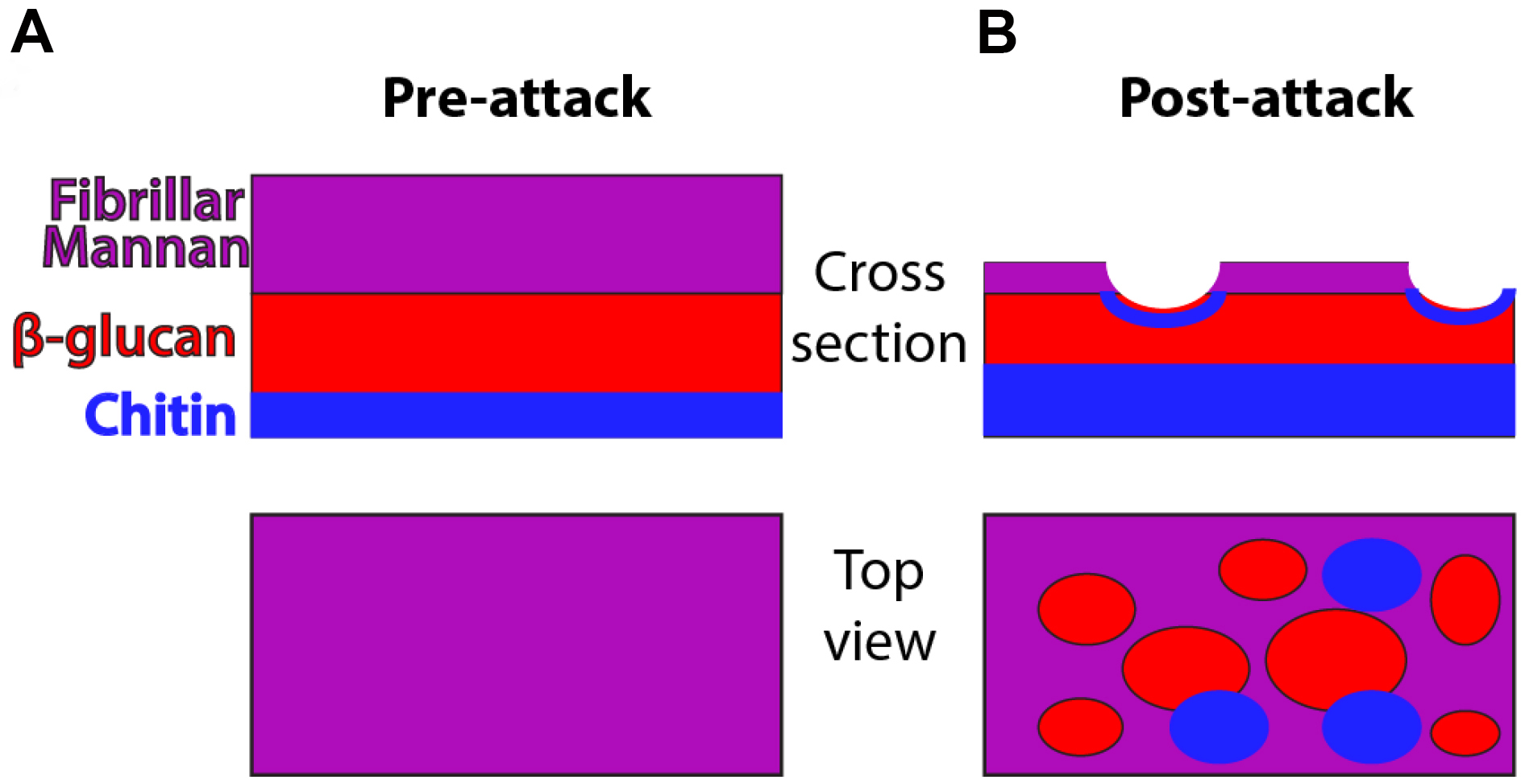
Figure 1. Schematic of cell wall organization pre- and post-neutrophil attack. Under homeostatic conditions, hyphal cell wall is composed of three main polysaccharide components (mannan, β-glucan and chitin) but most chitin and β-glucan are inaccessible for recognition because it lies beneath the mannan layer. Post-attack there is loss of cell wall mannoprotein and increased levels of chitin. These changes lead to greater surface recognition of β-glucan and perhaps also chitin.
Materials and Reagents
- Protective gloves and lab coat
- Flint glass culture tubes 16 x 150 mm (VWR, catalog number: 60825-435 )
- 50 ml conical tubes (Corning, Falcon®, catalog number: 352098 )
- Fisherbrand 100 x 20 mm Petri dishes (Fisher Scientific, catalog number: FB0875711Z )
- 10 ml syringe (BD, catalog number: 301604 )
- 25 G 5/8 needle (BD, catalog number: 305122 )
- 70 µm cell strainers (Corning, Falcon®, catalog number: 352350 )
- 1.7 ml microcentrifuge tubes
- Plain microscope slides (VWR, catalog number: 48300-025 )
- Kim-KapTM Disposable closures; 16 mm (Kimble Chase Life Science and Research Products, catalog number: 7366316 )
- Miltenyi columns (Miltenyi Biotec, catalog number: 130-021-101 )
- WT-FarRed670 strain of C. albicans (Hopke et al., 2016)
- WT-GFP strain (Wheeler et al., 2008)
- C57BL/6J mice, female 6-10 weeks old (THE JACKSON LABORATORIES, catalog number: 000664 )
- Glycerol (EMD Millipore, catalog number: GX0185-5 )
- Anti-Ly6G biotin antibody (Thermo Fisher Scientific, eBioscience, catalog number: 13-5931-85 )
- Anti-biotin magnetic beads (Miltenyi Biotec, catalog number: 130-090-485 )
- Trypan blue (Lonza, catalog number: 17-942E )
- FluoReporter® Cell-Surface Biotinylation Kit (Thermo Fisher Scientific, Molecular ProbesTM, catalog number: F20650 )
- Alexa Fluor 647 conjugated streptavidin (Jackson ImmunoResearch, catalog number: 016-600-084 )
- Anti-myeloperoxidase (MPO) (R&D Systems, catalog number: AF3667 )
- Anti-histone H3 (citrulline R2+R8+R17) (Abcam, catalog number: ab5103 )
- Sytox Green (Thermo Fisher Scientific, Molecular ProbesTM, catalog number: S7020 )
- Calcofluor white/Fluorescent brightener 28 (Sigma-Aldrich, catalog number: F3543 )
- Donkey anti-goat IgG Cy3 (Jackson ImmunoResearch, catalog number: 705-165-147 )
- Donkey anti-rabbit IgG Cy3 (Jackson ImmunoResearch, catalog number: 711-165-152 )
- sDectin-1-Fc (produced in house according to [Graham et al., 2006])
- Donkey anti-human IgG Cy3 (Jackson ImmunoResearch, catalog number: 709-165-149 )
- Nail polish
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: S271-3 )
- Sodium phosphate, dibasic anhydrous (Na2HPO4) (Fisher Scientific, catalog number: S374-500 )
- Potassium phosphate, monobasic anhydrous (KH2PO4) (Fisher Scientific, catalog number: P285-500 )
- Sodium carbonate (Na2CO3) (Sigma-Aldrich, catalog number: S7795 )
- BD Bacto peptone (BD, BactoTM, catalog number: 211677 )
- BD Bacto yeast extract (BD, BactoTM, catalog number: 212750 )
- Dextrose (Fisher Scientific, catalog number: D-163 )
- BD Bacto agar (BD, BactoTM, catalog number: 214014 )
- Heat inactivated fetal bovine serum (Thermo Fisher Scientific, GibcoTM, catalog number: 10082147 )
- Probumin® bovine serum albumin diagnostic grade powder (EMD Millipore, catalog number: 820451 )
- RPMI, with 25 mM HEPES and L-glutamine (Lonza, catalog number: 12-115F )
- Ammonium chloride (NH4Cl) (EMD Millipore, catalog number: AX12701 )
- Crystal violet (Sigma-Aldrich, catalog number: C0775 )
- Acetic acid (Fisher Scientific, catalog number: A38-212 )
- Tris base (AMRESCO, catalog number: 0497 )
- Hydrochloric acid (HCl) (Fisher Scientific, catalog number: A144-500 )
- PBS, pH 7.2 (see Recipes)
- PBS + Na2CO3, pH 8 (see Recipes)
- YPD broth (see Recipes)
- PBS + 5% FBS (see Recipes)
- PBS + 2% FBS (see Recipes)
- PBS + 2% BSA (see Recipes)
- RPMI + 5% FBS (see Recipes)
- Tris-HCl, pH 8.0 (see Recipes)
- TAC buffer (see Recipes)
- Turks solution (see Recipes)
Equipment
- Roller drum (Eppendorf, New Brunswick Scientific) and incubators (VWR)
- Hemacytometer for cell counting (Hausser Scientific, catalog number: 1492 )
- Spectrophotometer (Eppendorf; Biophotometer)
- Centrifuge with adaptors for 15 and 50 ml conical tubes (Thermo Fisher Scientific, model: SorvallTM LegendTM RT )
- AutoMACs separation system (Miltenyi Biotec, model: autoMACS® Pro Separator )
- Zeiss Axiovision fluorescence microscope (Zeiss; custom build)
- Dissection kit (VWR, catalog number: 631-0616 )
- pH meter (Fisher Scientific, model: accumetTM Excel XL15 )
- Microcentrifuge for 1.7 ml tubes (Eppendorf, model: 5424 )
- Autoclave
- 500 ml bottle (VWR, catalog number: 89000-233 )
- Water bath
Software
- AxioVision Rel48 software (Zeiss: https://www.zeiss.com/microscopy/us/downloads/axiovision-downloads.html)
- Photoshop (Adobe Systems, San Jose, CA)
Procedure
- Growth of Candida albicans hyphae (3 days; see Figure 2A for a schematic of the procedure).
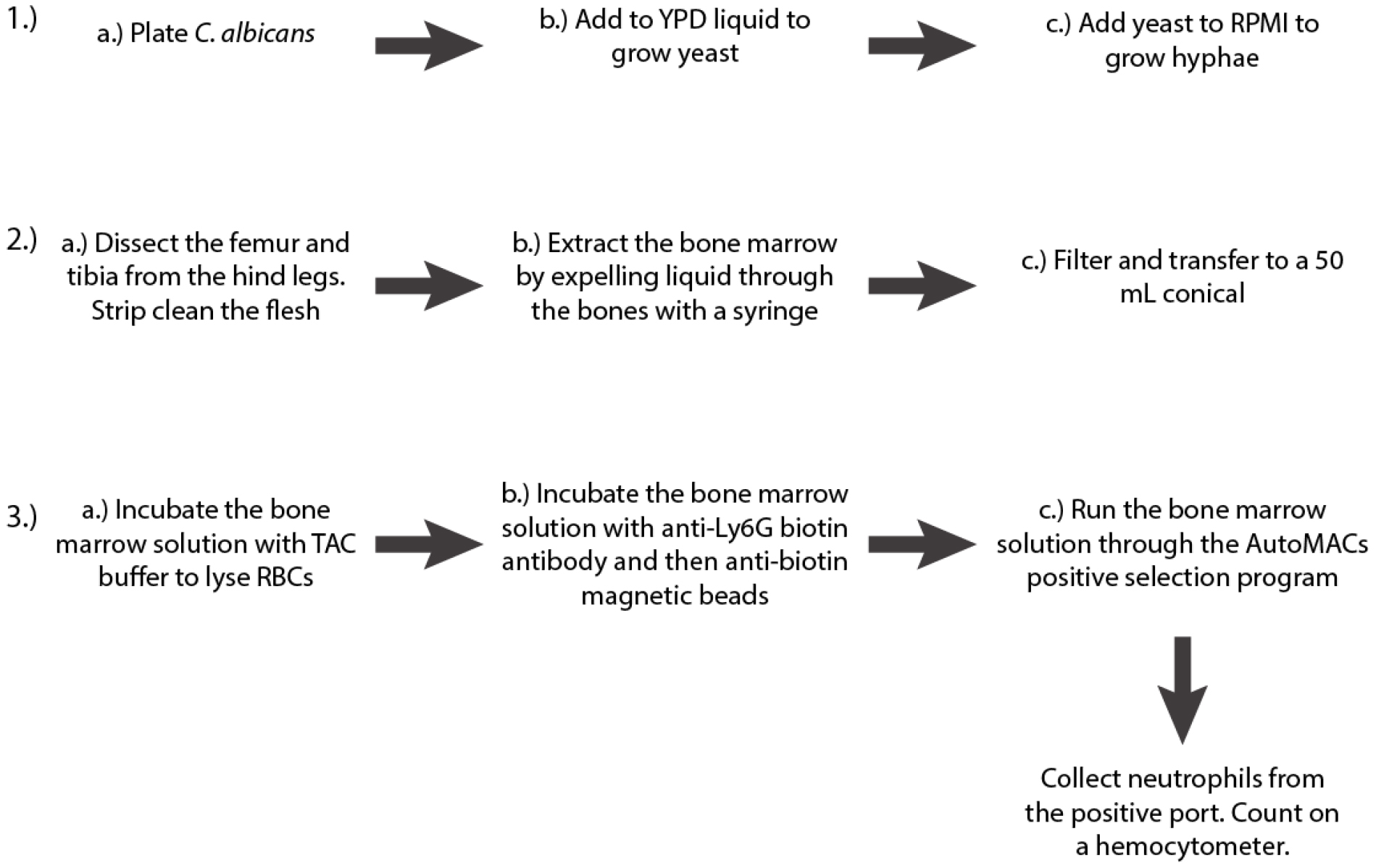
Figure 2. Schematic of the first three steps of the protocol - Streak the appropriate strain of C. albicans to single colonies from a frozen 25% glycerol stock onto YPD agar (see Recipes) and incubate at 37 °C overnight. For these experiments, we use the WT-FarRed670 strain of C. albicans (Genotype: PENO1::PENO1-FarRed670-NATR; [Hopke et al., 2016]).
- The next day pick a single colony and transfer into 5 ml of YPD liquid (see Recipes). We put the 5 ml in 16 x 150 mm flint glass tubes with caps and incubate at 30 °C on a roller drum overnight. Harvest the YPD culture and count the yeast using a hemocytometer.
- Dilute into 30 ml of RPMI at a concentration of 2.5 x 106 cells/ml and divide into 5 ml aliquots. Incubate at 30 °C on a roller drum overnight. Pool the RPMI cultures into a 50 ml conical tube and spin down at 3,000 x g for 5 min. Remove most of the supernatant (so only about 5 ml remains) and then flick to resuspend the pellet in remaining liquid. Due to the morphology of the hyphae, you cannot use a hemocytometer to determine cell number, instead we used the spectrophotometer to estimate the concentration of the hyphae. An OD600 of 1 is equivalent to 1 x 107 cells/ml. While quantification of hyphae by OD600 is not as accurate as for quantifying yeast, we found it to provide reproducible results. Adjust to 3 x 108 cells/ml to use as a stock solution in the assay.
Note: You will have a highly concentrated number of hyphae in a small volume, so it is best to do a dilution before finding the OD600 to ensure you fall within a readable range and to conserve cells. Our cuvettes usually take 1 ml, so we take 100 µl hyphae and add it to 900 µl PBS. PBS will then serve as your blank control. - Isolation of mouse bone marrow (1-2 h; see Figure 2B for a schematic of the procedure; for a detailed video protocol see [Swamydas and Lionakis, 2013])
- Euthanize two C57BL/6J female mice of 6-10 weeks of age via CO2 inhalation followed by cervical dislocation. Dissect the hind legs to acquire the femurs and tibiae. Remove all the flesh and transfer into a Petri dish with PBS + 5% FBS (see Recipes).
- Fill a 10 ml syringe with the PBS + 5% FBS and attach a 25 G 5/8 needle. Insert the needle into the bones and expel the liquid from the syringe to force out the bone marrow into the Petri dish (you can clip the ends of the bones if it is difficult to insert the needle).
- When all the bone marrow has been released into the Petri dish, pass it through a 70 µm cell strainer and into a 50 ml conical. Centrifuge at 300 x g for 3 min.
- Euthanize two C57BL/6J female mice of 6-10 weeks of age via CO2 inhalation followed by cervical dislocation. Dissect the hind legs to acquire the femurs and tibiae. Remove all the flesh and transfer into a Petri dish with PBS + 5% FBS (see Recipes).
- It is best to expel the bone marrow in a Petri dish that has no remnants of soft tissue. The tissue will clog the needle. If necessary, you can finish cleaning the bones in one Petri dish and then move the bones to another one with PBS + 5% FBS before expelling the marrow.
- All animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animals were euthanized by carbon dioxide inhalation. Infected animals were monitored twice daily for signs of infection and morbid animals were euthanized. The UMaine IACUC/Ethics Committee approved this protocol.
- Isolation of neutrophils from mouse bone marrow via AutoMACs (1.5-2 h; see Figure 2C for a schematic of the procedure)
- Discard the supernatant and resuspend the pellet by flicking the conical tube. Add 3 ml of Tris/ammonium chloride (TAC) buffer (see Recipes) and incubate at room temperature for 1 min to lyse any red blood cells. After the incubation, dilute to 30 ml total with PBS + 2% FBS. Take 10 µl of the bone marrow solution and add it to 40 µl Turks solution (see Recipes). Count the cells on a hemocytometer.
- Centrifuge the bone marrow solution at 300 x g for 3 min. Discard the supernatant and flick resuspend the pellet in PBS + 2% FBS at a concentration of 1 x 108 cells/ml. Incubate with anti-Ly6G biotin antibody (stock at 0.5 mg/ml) at 1 µl antibody per 100 µl bone marrow on ice for 15 min. Wash by centrifuging at 300 x g for 3 min and discarding the supernatant. Resuspend in 10 ml PBS + 5% FBS then centrifuge again. Discard the supernatant and resuspend again in 10 ml PBS + 5% FBS. Incubate with anti-biotin magnetic beads (10 µl/1 x 107 cells) on ice for 30 min. Agitate periodically throughout the incubation. Wash once with 10 ml PBS + 5% FBS as outlined above. Resuspend the pellet in 1 ml of PBS + 5% FBS.
Note: During this time turn on the AutoMACS and run the CLEAN cycle. - Once finished put clean 50 ml conical tubes in the positive and negative ports. Put the bone marrow at the intake port and run the positive selection program. When all but 0.2 ml of the volume has been taken up the intake, add 1 ml PBS + 2% FBS. This ensures that all of the residual sample is loaded onto the column without significantly increasing the volume. Collect both the positive and negative fractions*. Centrifuge the positive fraction at 300 x g for 3 min. Discard the supernatant and resuspend in 2 ml of RPMI + 5% FBS. Take 10 µl and add to 90 µl trypan blue before counting neutrophils on a hemocytometer.
- *The negative fraction is saved because it can be re-run if there is an issue with the machine and the neutrophils are not separated. It can be discarded once you have confirmed the presence of neutrophils in the positive fraction. Alternatively, the negative fraction can be used to generate bone marrow derived macrophages.
- Discard the supernatant and resuspend the pellet by flicking the conical tube. Add 3 ml of Tris/ammonium chloride (TAC) buffer (see Recipes) and incubate at room temperature for 1 min to lyse any red blood cells. After the incubation, dilute to 30 ml total with PBS + 2% FBS. Take 10 µl of the bone marrow solution and add it to 40 µl Turks solution (see Recipes). Count the cells on a hemocytometer.
- Turks solution is used to count cells in collected bone marrow because it is effective at lysing red blood cells that are not lysed by the TAC treatment. It is important to lyse red blood cells because it makes it much easier to count white blood cells.
- The expected yield from tibiae and femurs from two BL/6J mice is around 1 x 108 cells. The expected yield of neutrophils from two BL/6J mice is usually at least 1.4 x 107cells.
- Neutrophil purification is monitored by light microscopy during counting on the hemocytometer. Additional confirmation can come from labeling with anti-Ly6G antibodies and analysis with flow cytometry (see [Swamydas and Lionakis, 2013]).
- The details of this process will vary depending on the specific magnetic cell sorter. These details are for the AutoMACS (Miltenyi), although purification can be performed manually with Miltenyi columns instead.
- A video explaining clearly how to use the AutoMACS is available from the manufacturer. See the following URL: https://www.youtube.com/watch?v=h06klEgji4o.
- Biotinylation and fluorescent strepavidin labeling of C. albicans hyphal cell wall.
Note: To examine interaction of neutrophils with the cell wall, a fluorescent label can be used. This labeling can be done in parallel with the neutrophil separation from bone marrow. While we did not typically use this step in the experiments involved in looking at NETs, it was used extensively in other assays looking at neutrophil mediated changes to the fungal cell wall. A basic diagram of the C. albicans cell wall is shown in Figure 1A. Detailed schematics of C. albicans cell wall are widely available in the literature (see [Perez-Garcia et al., 2011]):- This protocol uses the FluoReporter® Cell-Surface Biotinylation Kit. In this kit, amine-reactive biotin bonds covalently with cell wall proteins. Using a new kit, take one biotin vial and resuspend it in 250 µl DMSO to make a 0.2 mg/ml stock solution of biotin-XX SSE. This can be stored at -20 °C and reused in later experiments until gone.
- With your C. albicans at 3 x 108 cells/ml, take enough volume so that you will have 3 x 107 cells for each sample you are going to run. To maintain this concentration, do all wash steps with the same volume of liquid and with 30 sec spins at 15,000 x g in a microcentrifuge.
- Wash once with PBS + Na2CO3 at pH 8 (see Recipes).
- Resuspend the hyphae in PBS + Na2CO3 (pH 8) and stain with biotin-XX SSE at 0.01 µg/µl for 15 min at room temperature.
- Wash three times with PBS pH 7.2. Resuspend in PBS pH 7.2.
- Stain with a fluorescently labeled streptavidin at 36 µg/ml for 30 min at room temperature.
- Wash once with PBS pH 7.2 and resuspend in PBS pH 7.2 until ready for use with neutrophils.
- This protocol uses the FluoReporter® Cell-Surface Biotinylation Kit. In this kit, amine-reactive biotin bonds covalently with cell wall proteins. Using a new kit, take one biotin vial and resuspend it in 250 µl DMSO to make a 0.2 mg/ml stock solution of biotin-XX SSE. This can be stored at -20 °C and reused in later experiments until gone.
- Hyphal/Immune cell interaction (2.5 h)
Incubate 3 x 107 hyphal cells with or without 7 x 106 neutrophils in 1.7 ml microcentrifuge tubes with RPMI + 5% FBS, in a total of 1.7 ml, on a rotator at 37 °C for 2.5 h. The sample without neutrophils serves as a control.
Note: It is important to have the microcentrifuge tube almost completely full of media so that neutrophils and hyphae do not get trapped and dried out on the sides of the tube in an air pocket. - Staining for NET visualization (3 h; for a schematic diagram of the staining process see Figure 3A)
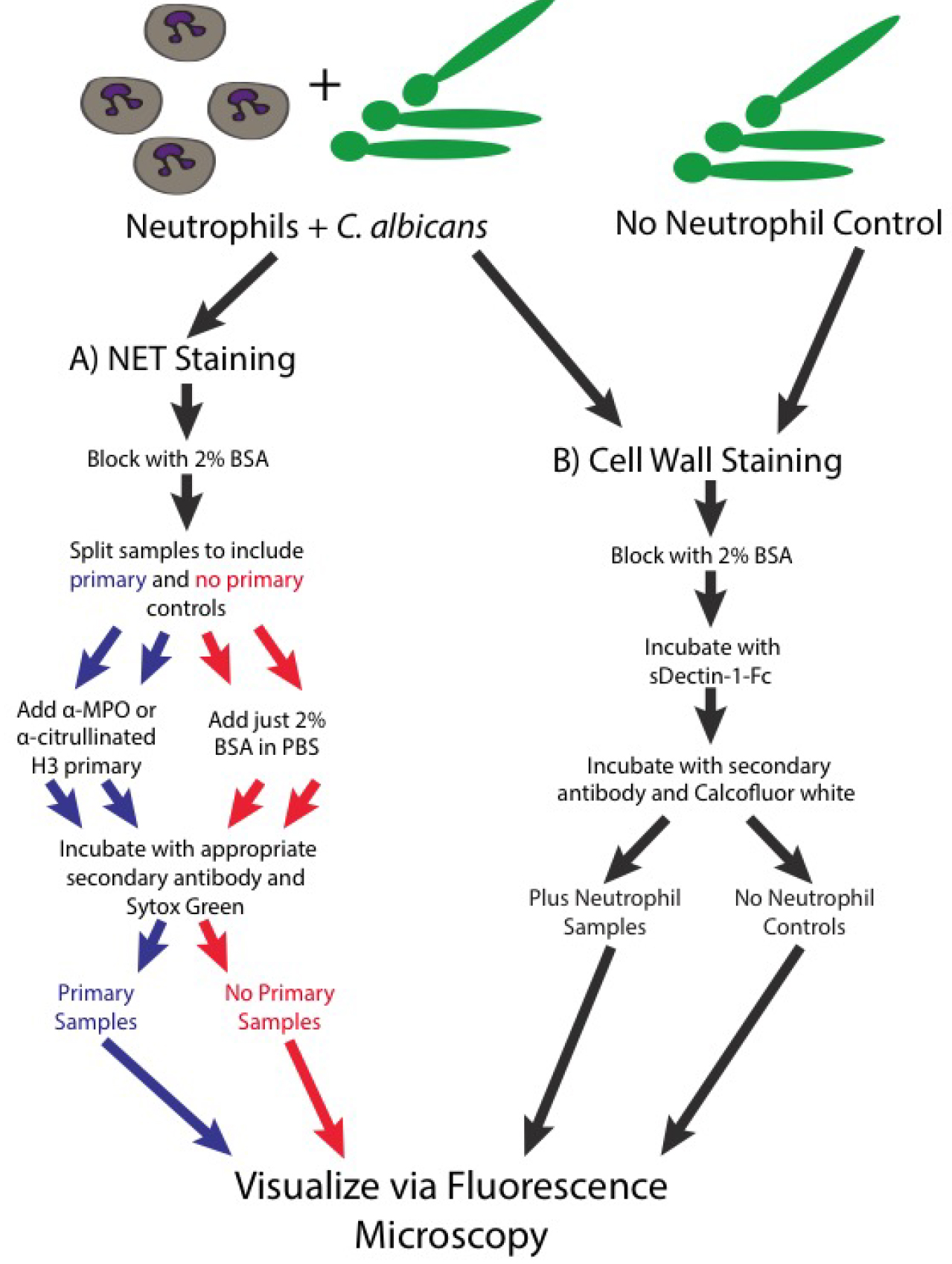
Figure 3. Schematic of the staining steps of the protocol- Note that all steps are done on ice and with ice-cold buffers, unless specified otherwise. This prevents any additional metabolic functions in these live cells.
- Spin down the samples in a microcentrifuge at max speed (15,000-21,000 x g) for 30 sec. Remove the supernatants and wash once with 500 µl of PBS.
- Resuspend in 200 µl of PBS + 2% BSA to block the samples. Incubate at room temperature for 30 min.
- At this point, split all the samples in half, one will receive primary antibody and the other will not to serve as a control.
- Spin samples in the microcentrifuge tube and discard the supernatant.
- Resuspend the samples in 100 µl PBS + 2% BSA with primary antibody or with just PBS + 2% BSA (no primary control). The primary antibody can either be anti-myeloperoxidase (0.1 mg/ml) or anti-histone H3 citrulline R2+R8+R17 (0.014 mg/ml).
- Incubate the samples on ice for 1 h.
- Wash the samples 3-5 times with 500 µl PBS.
- After the final wash, resuspend all samples (both the primary and the no primary controls) in a total of 100 µl PBS + 2% BSA with Sytox Green (156 nM), calcofluor white (25 ng/ml) and secondary antibody. For the samples stained with anti-MPO primary, the secondary antibody we used was donkey anti-goat IgG Cy3 (0.007 mg/ml) and for the samples stained with anti-histone, the secondary we used was donkey anti-rabbit IgG Cy3 (0.0075 mg/ml).
Note: Sytox Green is a non-membrane permeable DNA stain which will fluoresce brightly after binding the extracellular DNA found in NETs. CFW is a stain for chitin in the fungal cell wall. - Incubate for 30 min at room temperature.
- After incubation, wash the samples 3-5 times with PBS and resuspend in 35 µl of PBS.
- Note that all steps are done on ice and with ice-cold buffers, unless specified otherwise. This prevents any additional metabolic functions in these live cells.
- Visualizing neutrophil mediated disruptions of the fungal cell wall (for a schematic diagram of the staining process see Figure 3B)
- As an alternative to staining for NET components, a slight alteration of the protocol will allow the visualization of neutrophil mediated changes to the fungal cell wall. This involves staining with sDectin-1-Fc to look for the exposure of the inflammatory fungal pathogen associated molecular pattern β-glucan, staining with calcofluor white to look for changes in fungal chitin and the fluorescent labeling of the outer cell wall as outlined in step 4 to examine immune mediated disruptions of this outer layer. Steps 1-5 are followed as outlined above but step 6 is skipped.
- Spin down the samples in a microcentrifuge at max speed for 30 sec. Remove the supernatants and wash once with 500 µl of PBS.
- Resuspend in 200 µl of PBS + 2% BSA to block the samples. Incubate at room temperature for 30 min.
- Resuspend the samples in a total of 50 µl PBS + 2% BSA with sDectin-1-Fc at 17 µg/ml. Incubate on ice for 1 h.
- Wash the samples 3-5 times with 500 µl PBS.
- After the final wash, resuspend all samples in a total of 200 µl of PBS + 2% BSA with calcofluor white (CFW; 25 ng/ml) and donkey anti-human IgG Cy3 secondary antibody (0.8 mg/ml). Incubate for 30 min at room temperature. After incubation, wash the samples 3-5 times with PBS and resuspend in 35 µl of PBS.
Note: In order to reduce the amount of sDectin-1-Fc used per sample, the sDectin-1-Fc staining step is typically done in a smaller volume (50 µl) than the blocking or secondary antibody steps which are done with 200 µl. - The samples are now ready to visualize by fluorescence microscopy. Using clean glass slides, load 5 µl of sample and place a coverslip on top. The coverslip can be sealed with nail polish to prevent drying out during imaging. Depending on the experimental question, random fields of view or specific sites were chosen and imaged.
- Once set, the exposure times for all channels were kept the same for all images. An equal number of images were obtained from all samples for use in analysis.
- The hyphae will have the chitin in their cell walls stained with the CFW. Areas of exposed β-glucan that is bound by sDectin-1-Fc will be marked by Cy3 fluorescent signal (shown via diagram in Figure 1B and via microscopy in Figure 4). Areas of neutrophil attack of the outer cell wall can be detected by examining the loss of fluorescent streptavidin signal (Figure 4A). A viability indicator is usually included by using a C. albicans strain with cytoplasmic fluorophore expression. For these experiments, a prototrophic WT-GFP strain (Genotype: SC5314 Peno1::Peno1-EGFP-NATR; [Wheeler et al., 2008]) was typically used.
Data analysis
- For NET staining, analysis focused on the qualitative evaluation of whether MPO and citrullinated H3 signals were present in the stained samples, co-localized with extracellular DNA and did not appear in the no primary controls (Figure 5).
- For analysis of cell wall damage, a number of options are available and depend on the hypothesis being tested. For images with z-stacks, we first create maximum image projections using the standard Zeiss Axiovision software (Zeiss Microscopy). One way we have analyzed these data is by examining the percentage of viable cells in all images which display specific cell wall changes (Figure 4D). This type of analysis is useful when attempting to determine if specific inhibitors or genetic knockouts, on either the fungal or neutrophil side, disrupt the process. We have also analyzed this data by determining the mean fluorescent intensity (MFI) of the staining at specific sites of interest and comparing this to the MFIs seen in controls (Figure 4C). The MFI can be obtained with numerous programs. We captured our images with a Zeiss Axiovision microscope and determined the MFI with Axiovision software. This analysis has proven useful in quantitatively measuring the overlap of specific cell wall changes (for example: increased sDectin-1-Fc staining overlaps at areas of increased CFW staining at neutrophil attack sites). It has also proven useful in demonstrating the changes in levels of cell wall components at specific sites when using fungal strains with fluorescent protein fusions.
Notes:
- In order to do these comparisons, all images being compared within an experiment (experimental samples and controls) must be acquired with the same exposure times. An equal number of images for all samples should also be obtained and analyzed. We usually use between 12 and 20 fields of view per sample per experiment. These fields of view can be obtained randomly or by finding specific sites depending on the experiment; note that fields should be chosen using a neutral channel (such as brightfield) to eliminate potential bias. Typically, three independent experiments were performed and data are shown as the average of the three experiments ± standard error. Note that here quantification of only a single field is shown so there are no error bars.
- When analyzing images, we avoid including the tips of hyphae in the analysis. These tips can be the sites of new growth that were not originally biotinylated and this growth can cause cell wall changes unrelated to the interaction with neutrophils that would complicate the analysis. Examples of these areas are shown in Figures 4E-4F. We also avoid hyphae that are overlapping each other, especially when examining MFI, to make sure each hyphal segment is well resolved.
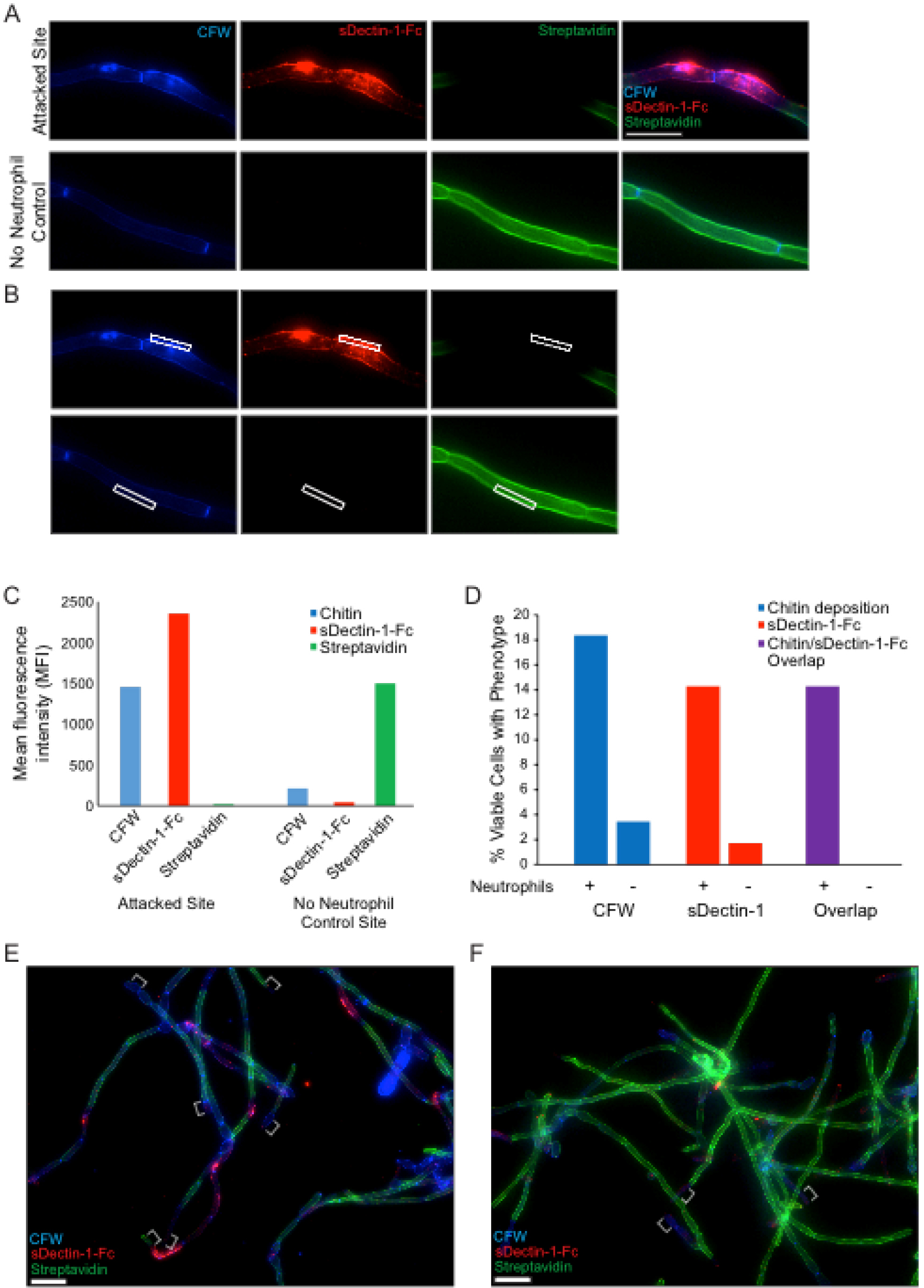
Figure 4. In vitro staining of hyphal cell wall after neutrophil attack. Representative results for the sDectin-1-Fc staining assay are shown for an attack site and for the no neutrophil control. Panel A illustrates typical staining of both a site attacked by a neutrophil (top) and by the normal unattacked cell wall in a sample of C. albicans hyphae not challenged with neutrophils (bottom). Panel B shows how fluorescence intensity of different labels is measured using a region-of-interest box in Axiovision software to obtain the mean fluorescence intensity (MFI) for each channel. The data comparing fluorescence intensity between a single attack site and a no neutrophil control site are shown in panel C. In addition to quantitative measurement of fluorescence intensities, the percentage of cells displaying a given phenotype (over a number of fields imaged) is also quantified. Panel D shows representative results from quantification of two single fields of view shown in panels E and F. Examples of hyphal tips, which were avoided during analysis, are indicated with white brackets (E-F). More extensive analysis can be seen in Hopke et al., 2016. Scale bar represents 10 µm (A) or 20 µm (E-F).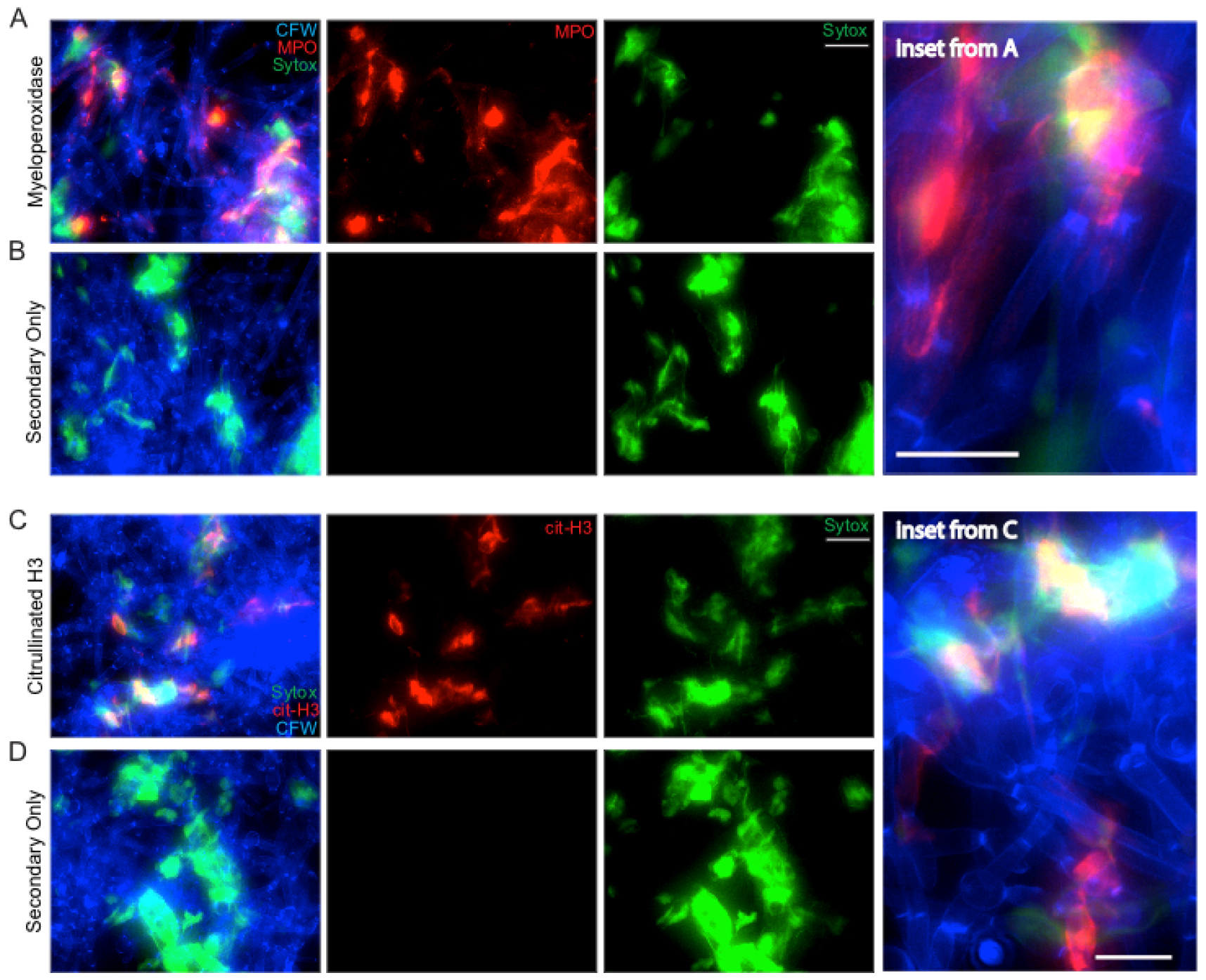
Figure 5. NET staining assay. Representative results for the NET staining assay are shown for both MPO (A & inset) and citrullinated histone H3 (C & inset). The corresponding secondary antibody only controls are shown below each (B and D). Panels shown include the individual channels for Sytox green staining and either MPO or citrullinated H3 staining as well as the overlay of these channels. Scale bar represents 10 µm (insets) or 20 µm (A-B, C-D).
- Representative results
Representative staining results for NET components are shown in Figure 5. C. albicans hyphae staining by CFW is shown in blue. Extracellular DNA staining by Sytox is shown in green. Staining with MPO (Figures 5A-5B) or citrullinated H3 histone antibody (Figures 5C-5D) is shown in red. The no primary (NP) controls show a lack of any signal in the red channel, demonstrating the specificity of the primary antibody during the staining process (Figures 5B and 5D). MPO staining signal co-localizes with extracellular DNA but also directly on hyphae, suggesting MPO is released by neutrophils during contact with hyphae both within and beyond the context of NETs (Figure 5A, inset). Citrullinated histone H3 signal localizes with extracellular DNA staining only, suggesting it is only released by neutrophils in the context of NETs and stays closely bound to DNA (Figure 5C, inset). Co-localization of staining with all three classical NET markers was used to demonstrate true NETs were being formed during incubation of neutrophils with hyphae.
Representative results for staining to examine fungal cell wall damage are shown in Figure 4A neutrophil attack site is shown, with increased CFW staining, strong sDectin-1-Fc staining and a loss of Alexa Fluor 647-conjugated Streptavidin signal (Figure 4A). A site from the no neutrophil control sample is also shown for comparison. These controls show the phenotype of the normal hyphal cell wall. Examples of data analysis from experiments done with this protocol are also shown including the mean fluorescence intensity (MFI) for different cell wall stains at both the attack and control site (Figures 4B-4C) and the percentage of viable cells showing cell wall changes (Figure 4D) from example images (Figures 4E-4F).
Recipes
- PBS, pH 7.2
154 mM sodium chloride
5.6 mM sodium phosphate, dibasic anhydrous
1.06 mM potassium phosphate, monobasic anhydrous
Adjust pH to 7.2
Autoclave for 20 min on liquids cycle - PBS + Na2CO3, pH 8
Prepare a 1 M solution of Na2CO3, pH 10.5
Add 1/10 volume to PBS to increase pH to ~8 to enhance amine-reactive chemistry - YPD broth
20 g/L BD Bacto peptone
10 g/L BD Bacto yeast extract
20 g/L dextrose
Fill to 1 L with distilled water
Autoclave at 121 °C for 20 min on liquids cycle
Note: YPD agar is the same as above with the addition of 20 g/L BD Bacto agar. After autoclaving, wait for the bottle to cool to the point it can be handled and make agar plates by adding 25 ml molten agar to 100 x 20 mm Petri dishes. Store at 4 °C until ready to use. - PBS + 5% FBS
47.5 ml PBS
2.5 ml heat inactivated fetal bovine serum
Note: We typically heat-inactivate FBS by placing a 500 ml bottle in a 55 °C water bath for 20 min. We then pre-aliquot the FBS by adding 2.5 ml to 50 ml conicals and freezing them at -20 °C. They can then be thawed when ready to use. Making up two tubes of this is usually enough for one experiment. - PBS + 2% FBS
24 ml PBS
16 ml PBS + 5% FBS
Note: You can make up the PBS + 2% FBS directly or you can dilute some of the PBS + 5% FBS that you will have already made as outlined above. - PBS + 2% BSA
50 ml PBS
1 g Probumin® bovine serum albumin diagnostic grade powder
Note: The PBS + 2% BSA is stored at 4 °C until just before use. Be sure to warm it up to room temperature before opening cap to prevent condensation of moisture inside bottle. - RPMI + 5% FBS
RPMI 1640, with 25 mM HEPES and L-glutamine
Heat inactivated fetal bovine serum
Note: This solution is made up fresh each day for the experiment. The amount needed will depend on the number of samples you are running that day. - Tris-HCl, pH 8.0
1 M Tris base
Adjust pH to 8.0 with concentrated hydrochloric acid - TAC buffer (3 ml for 1 min)
0.017 M Tris-HCl, pH 8.0
0.114 M NH4Cl - Turks solution
0.01% crystal violet
3% acetic acid
Acknowledgments
This work has been supported by grants from the following sources: the U.S. Department of Agriculture (ME0-H-1-00517-13), the National Institutes of Health (R15AI094406), and the Burroughs Wellcome Fund to Dr. Robert T. Wheeler. This manuscript is Maine Agricultural and Forest Experiment Station Publication Number 3481.
References
- Amulic, B., Cazalet, C., Hayes, G.L., Metzler, K.D., Zychlinsky, A. (2012). Neutrophil function: from mechanisms to disease. Annu Rev Immunol (30): 459-489.
- Branzk, N., Lubojemska, A., Hardison, S. E., Wang, Q., Gutierrez, M. G., Brown, G. D. and Papayannopoulos, V. (2014). Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 15(11): 1017-1025.
- Branzk, N. and Papayannopoulos, V. (2013). Molecular mechanisms regulating NETosis in infection and disease. Semin immunopathol (35): 513-530.
- Brown, G. D., Denning, D. W., Gow, N. A., Levitz, S. M., Netea, M. G. and White, T. C. (2012). Hidden killers: human fungal infections. Sci Transl Med 4(165): 165rv113.
- Bruns, S., Kniemeyer, O., Hasenberg, M., Aimanianda, V., Nietzsche, S., Thywissen, A., Jeron, A., Latge, J. P., Brakhage, A. A. and Gunzer, M. (2010). Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog 6(4): e1000873.
- Graham, L. M., Tsoni, S. V., Willment, J. A., Williams, D. L., Taylor, P. R., Gordon, S., Dennehy, K. and Brown, G. D. (2006). Soluble Dectin-1 as a tool to detect beta-glucans. J Immunol Methods 314(1-2): 164-169.
- Hopke, A., Nicke, N., Hidu, E. E., Degani, G., Popolo, L. and Wheeler, R. T. (2016). Neutrophil attack triggers extracellular trap-dependent candida cell wall remodeling and altered immune recognition. PLoS Pathog 12(5): e1005644.
- Lionakis, M. S. and Netea, M. G. (2013). Candida and host determinants of susceptibility to invasive candidiasis. PLoS Pathog 9(1): e1003079.
- Perez-Garcia, L. A., Diaz-Jimenez, D. F., Lopez-Esparza, A. and Mora-Montes, H. M. (2011). Role of cell wall polysaccharides during recognition of Candida albicans by the innate immune system. Glycobiology 1.
- Rohm, M., Grimm, M. J., D’Auria, A. C., Almyroudis, N. G., Segal, B. H. and Urban, C. F. (2014). NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect Immun 82(5): 1766-1777.
- Swamydas, M. and Lionakis, M. S. (2013). Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp (77): e50586.
- Urban, C. F., Reichard, U., Brinkmann, V. and Zychlinsky, A. (2006). Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol 8(4): 668-676.
- Wheeler, R. T., Kombe, D., Agarwala, S. D. and Fink, G. R. (2008). Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog 4(12): e1000227.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Hopke, A. and Wheeler, R. T. (2017). In vitro Detection of Neutrophil Traps and Post-attack Cell Wall Changes in Candida Hyphae. Bio-protocol 7(7): e2213. DOI: 10.21769/BioProtoc.2213.
- Hopke, A., Nicke, N., Hidu, E. E., Degani, G., Popolo, L. and Wheeler, R. T. (2016). Neutrophil attack triggers extracellular trap-dependent candida cell wall remodeling and altered immune recognition. PLoS Pathog 12(5): e1005644.
Category
Immunology > Immune cell imaging > Confocal microscopy
Microbiology > Microbe-host interactions > Fungus
Cell Biology > Cell staining > Cell wall
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.