- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Wheat Root-dip Inoculation with Fusarium graminearum and Assessment of Root Rot Disease Severity


Published: Vol 7, Iss 6, Mar 20, 2017 DOI: 10.21769/BioProtoc.2189 Views: 12029
Reviewed by: Zhaohui LiuSwetha ReddyAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Fusarium graminearum is one of the most common and potent fungal pathogens of wheat (Triticum aestivum) and other cereals, known for causing devastating yield losses and mycotoxin contaminations of food and feed. The pathogen is mainly considered as a paradigm for the floral disease Fusarium head blight, while its ability to colonize wheat plants via root infection has been examined recently. F. graminearum has a unique infection strategy which comprises complex, specialized structures and processes. Root colonisation negatively affects plant development and leads to systemic plant invasion by tissue-adapted fungal strategies. The pathosystem wheat root - F. graminearum makes available an array of research areas, such as (i) the relatively unknown root interactions with a necrotrophic pathogen; (ii) genes and pathways contributing to (overall) Fusarium resistance; (iii) induced systemic (whole-plant) resistance; (iv) pathogenic strategies in a variety of host tissues; and (v) age-related changes in the single-genotype responses to seedling and adult plant (root/spike) infection. The presented Fusarium root rot bioassay allows for efficient infection of wheat roots, evaluation of disease severity and progress as well as statistical analysis of disease dynamics.
Keywords: Fusarium root rotBackground
The Fusarium root rot (FRR) bioassay uses root-dipping for inoculation in combination with different measurements of disease severity parameter. This protocol is principally also applicable to investigations of other root-fungus interactions. The presented root-dip inoculation proved to be an effective and reliable method to investigate wheat root-Fusariuminteractions (phenotypically and histologically) and to screen wheat genotypes for their response to root infection (Wang et al., 2015). Using the described protocol, genetic, molecular, and metabolomic aspects of the FRR disease have meanwhile been examined with reliable results in terms of biological repetitions and consistent observations across different research approaches. This corresponds with observations made in a study on the Verticillium wilt disease, which characterised root-dipping as superior to the pot immersion or soil infestation method in terms of effectiveness and reliability (Trapero et al., 2013). Growing wheat seedlings in F. graminearum contaminated (root zone) soil led to FRR-genotype responses similar to root dip inoculation (Wang et al., 2015), but the infection conditions are comparatively less controlled in terms of root specificity and time of infection. This might be a restriction for investigations that require time-based analyses. The use of Petri dishes to germinate and inoculate roots via mycelial agar plugs is a method that has been applied to F. culmorum infection in wheat seedlings (Beccari et al., 2011). However, in comparison to root dip inoculation, this method is not applicable to adult plant root infection, as was done in our lab to study plant age-related effects on FRR disease progress and wheat responses.
In the described protocol, disease severity can be assessed by percentage reductions of diseased root biomass, root and shoot length as well as by rates of visible root necrosis. FRR significantly inhibits root biomass production of wheat seedlings and adult plants (Wang et al., 2015), which can be measured by quantitative real-time PCR (qPCR). Disease severity and progress in terms of fungal growth can be monitored by measuring the relative amount of F. graminearum DNA in the host tissue by qPCR. This also enables detection and monitoring of infection during or in case of symptom-free disease periods. The F. graminearum spread into the lower stem internode is a crucial event as it initiates the colonisation of upper stem internodes, leaves, further tillers and even spikes (Wang et al., 2015) and can be readily evaluated by appearance time and rate of visual necrosis. For the FRR disease progress over time, a good agreement was found between the quantified relative F. graminearum biomass in roots and the measured impacts on seedling growth or the rated visible symptoms (Wang et al., 2015). Briefly, seedlings with the lowest level of F. graminearum accumulation measured displayed relative minor root necrosis and reductions in root biomass and length, while relatively moderate and maximum levels of pathogen accumulation each led to correspondingly moderated and maximum disease impacts and symptoms. Finally, Fusarium resistance is quantitative or partial. Therefore, the combination of classical, subjective tools such as symptom rating with the sensitive, non-subjective qPCR diagnosis of pathogen and/or root biomass proved to be advantageous, in terms of an improved assessment of disease dynamics and genotype performances.
Materials and Reagents
- Parafilm
- Cheese cloth
- Fine sand (washed and sieved), obtained from construction or agricultural market, autoclaved at 120 °C for 30 min
- Aluminium foil
- Polystyrene disk (thickness 10 mm)
- F. graminearum isolate ‘IFA 65’ (University of Natural Resources and Applied Life Sciences, Department for Agrobiotechnology, Vienna, Austria)
- Synthetic nutrient agar medium ‘Spezieller Nährstoffarmer Agar (SNA)’ (Leslie and Summerell, 2006)
- Tween-20 (Carl Roth, catalog number: 9127 )
- MENNO Florades (MENNO CHEMIE Norderstedt)
- Sodium hypochlorite solution (Carl Roth, catalog number: 9062 )
- Wuxal Super (Manna, Düsseldorf)
- Liquid nitrogen
- Potato dextrose broth (PDB) (Sigma-Aldrich, catalog number: P6685 )
- FastStart Universal SYBR Green Master (Roche Molecular Systems, catalog number: 04913850001 or 04913914001 )
- Potassium dihydrogen phosphate (KH2PO4)
- Potassium nitrate (KNO3)
- Magnesium chloride heptahydrate (MgSO4·7H2O)
- Potassium chloride (KCl)
- Glucose
- Sucrose
- Agar
- Synthetic nutrient deficient agar (SNA) (see Recipes)
Equipment
- Climate chamber with 20 °C under cool-white and near-UV light illumination for preparation of fungal culture
- Haemocytometer
- Light microscope (Zeiss)
- Magnetic stirrer
- Climate chamber with a 16 h photoperiod of 22 °C/18 °C day/night and 60% humidity for plant cultivation
- Flat tray
- Rotary shaker
- Pot (7.5 x 7.5 x 8.0 cm)
- NanoDrop ND 1000 (Thermo Fisher Scientific, model: NanoDrop ND 1000 )
- ABI Step One Plus real-time PCR system (Applied Biosystems)
Software
- PSS 20 (IBM SPSS Statistics 20; IBM Corp., USA)
Procedure
- Preparation of F. graminearum macroconidia suspension
- Store macroconidia suspension at -80 °C in sterile, double-distilled water as stock solution.
- To prepare macroconidia suspension for inoculation, transfer 15 µl of stock solution on a synthetic nutrient deficient agar (SNA) and culture at 20 °C under cool-white and near-UV light illumination. Use Parafilm to seal the agar plates. Please also take into consideration Note 1 on fungal aggressiveness given at the end of protocol.
- The 9-day-old fungal colony is ready for harvest of macroconidia. SNA is a weak nutrient agar. Hence, colonies grow slow and produce relatively low amounts of (fluffy) mycelia, which at harvest time vary from white to light pink in colour (Figure 1). Details on the macroconidia morphology are given by Leslie and Summerell (2006).
- Wash the SNA agar medium with 4 ml 0.02% (v/v) Tween-20 solution (diluted in sterile, double-distilled water) and then pass through four layers of sterile cheese cloth. Determine the concentration of macroconidia suspension by using a haemocytometer and adjust the concentration to 5 x 104 macroconidia/ml with 0.02% (v/v) Tween-20.
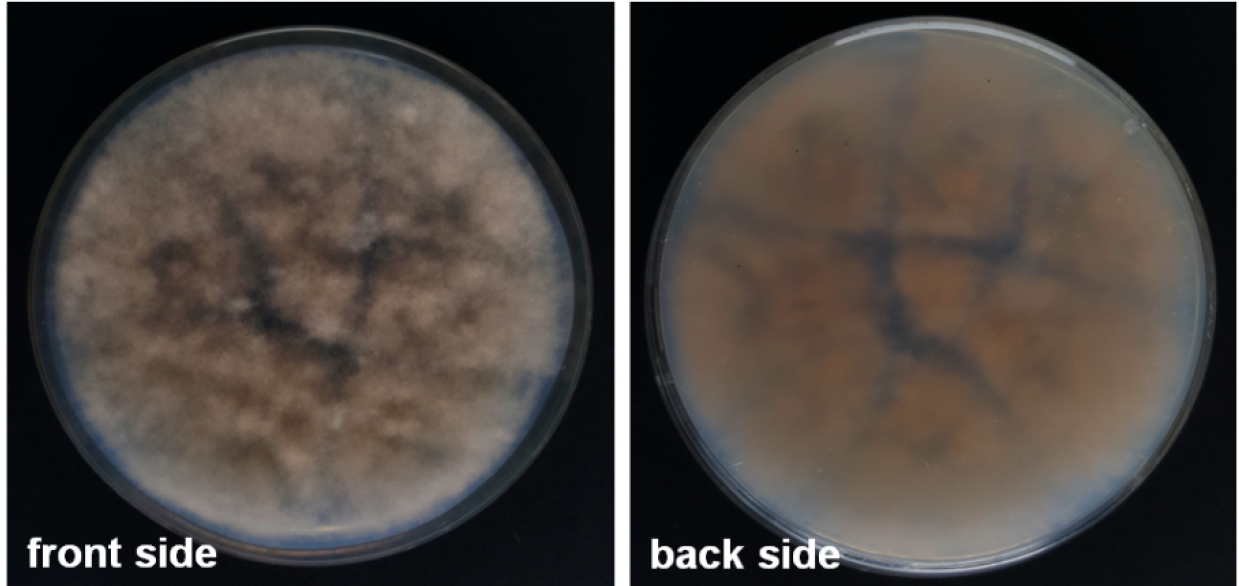
Figure 1. Fusarium graminearum colony on SNA plate at day of harvest
- Store macroconidia suspension at -80 °C in sterile, double-distilled water as stock solution.
- Root-dip inoculation [at seedling stages]
- Keep seedlings free from contamination. Clean the chamber prior to cultivation and inoculation by using an antimicrobial disinfecting agent, according to the manufacturer’s specifications [in this case with MENNO Florades].
- Sterilize seeds in sodium hypochlorite (6%) for 40 min on a magnetic stirrer; then wash 10 times with sterile double-distilled water.
- Sow seeds in autoclaved sand and cultivate plants in a climate chamber with a 16 h photoperiod of 22 °C/18 °C day/night and 60% humidity until the first leaf is unfolded [Zadoks (Z) 11; Zadoks et al., 1974].
- Prior to inoculation, remove plants carefully from the sand.
- Pool every five seedlings by wrapping their hypocotyls and stems with aluminium foil. This will ensure that only roots come in contact with the fungal suspension and allows fixation of seedlings as shown in Figure 2. In addition, seedlings can be held in place by using a slot in a polystyrene plug. Plugs are cut out from a polystyrene disk (thickness 10 mm) to the size of flat tray. For each chamber a hole is punched (a conical point chisel is useful), large enough to let pass seedling roots.
- For inoculation, place pooled seedlings into a flat tray (divided into chambers) by submerging their roots in 5 ml of macroconidia suspension (Figure 2); then shake gently for 2 h on a rotary shaker (Figure 2). For non-inoculated control plants, perform mock inoculation with 0.02% (v/v) Tween-20 (diluted in sterile, double-distilled water).

Figure 2. Root-dip inoculation of wheat seedlings with F. graminearum. Seedlings are placed into a small flat tray with 12 chambers each filled with 5 ml of macroconidia suspension. Hypocotyls and stems above the roots are wrapped in aluminium foil to avoid contact with the fungal suspension. - After inoculation, transplant a single or at maximum five seedlings (not closely packed together) in a single pot (7.5 x 7.5 x 8.0 cm) with autoclaved sand. Cultivate seedlings in a climate chamber set at 20 °C under a 22 °C/18 °C day/night rhythm with approximately 60% relative humidity. Microscopic examinations have shown that F. graminearum actively penetrates the epidermal layer of roots by forming different specialised infection structures. To avoid disruption of the infection process, transplant the seedlings immediately after removal from fungal solution. No further treatment is needed. Add 100 ml of water after planting. The water should be poured slowly and carefully.
- During cultivation the watering (250 ml per pot) regime dependents on the present growth chamber (greenhouse) conditions. General rule: Sand has a limited water holding capacity. Hence, we suggest to water as soon as the sand surface is dry [in this case every 4 to 6 days]. Please also take into consideration the Notes 2 and 3 on plant cultivation and experimental setup given at the end of protocol.
- Keep seedlings free from contamination. Clean the chamber prior to cultivation and inoculation by using an antimicrobial disinfecting agent, according to the manufacturer’s specifications [in this case with MENNO Florades].
- Disease severity assessment
- Tissue sampling
Collect (e.g., each 10) plants per sampling timepoint, treatment and genotype. Wash the roots under running water or immersed in water to avoid root injury. Roots should be washed until sand grains are removed from root surface. The parenchyma tissue of leaf sheath (wrapped around the young stem) is used by the pathogen to colonise lower stem. Hence, caution should be taken with washing, because diseased leaf sheath occasionally becomes very soft and drop gently. - Disease impacts on plant development
Measure the root and shoot length of inoculated and non-inoculated control plants. Calculate the relative reduction (percentage change) as the ratio between trait expression of diseased plants and trait expression of control plants: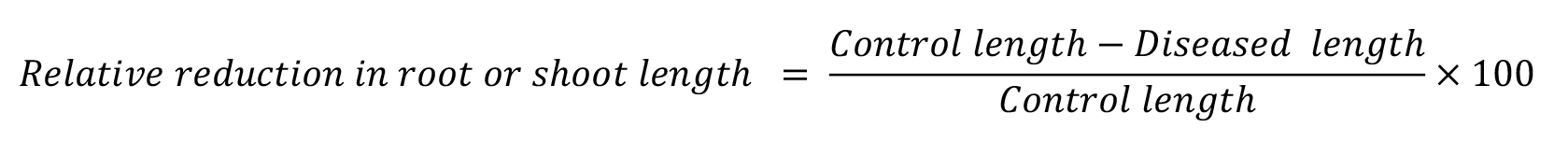
- Visible disease symptoms
Rate the symptoms on root and stem base by applying each browning and symptom extension scales (0 to 4) to single plants (Figure 3). The browning scale ranges from 0, symptomless; 1, slightly necrotic; 2, moderately necrotic; 3, severely necrotic; 4, completely necrotic; and the extension scale ranges from: 0, no lesions; 1, 1-24%; 2, 24-49%; 3, 50-75%; 4, > 75% discoloration of root or stem base. Calculate a genotype-timepoint-specific root and stem base symptom index (RSI and SbSI) by using the equation:
RSI or SbSI = (E1 + E2 + … + En/n) + (B1 + B2 + … + Bn/n)
Where,
B and E each represent the parameter browning and extension scale,
n represents the number of assessed individuals per genotype and treatment. Stem base is referred to the transition area between the root and the shoot system, located between sub-crown and first stem internode (Figure 3A).
An overview on different degrees of diseased root phenotypes and necrosis symptoms is given by Wang et al. (2015). In this publication, Supplementary Figure S1 shows root phenotypes of four wheat genotypes, representing resistant and moderate to high susceptible responses. On the roots and stem bases of susceptible genotypes symptoms typically show a transition from initially local, light (amber) brown discolorations to more distributed dark brown lesions at later timepoints. Stem base necrosis typically covers about half of the stem base of initially few individuals and continued until nearly the entire stem base was affected for more or less all individual plants. Please also take into consideration the Note 4 on symptom assessment given at the end of protocol.
Note: Create top view photos showing inoculated and non-inoculated plants side by side (Figure 3A). Such photos can, for example, be used for accurate measurement of browning extension by image analysis.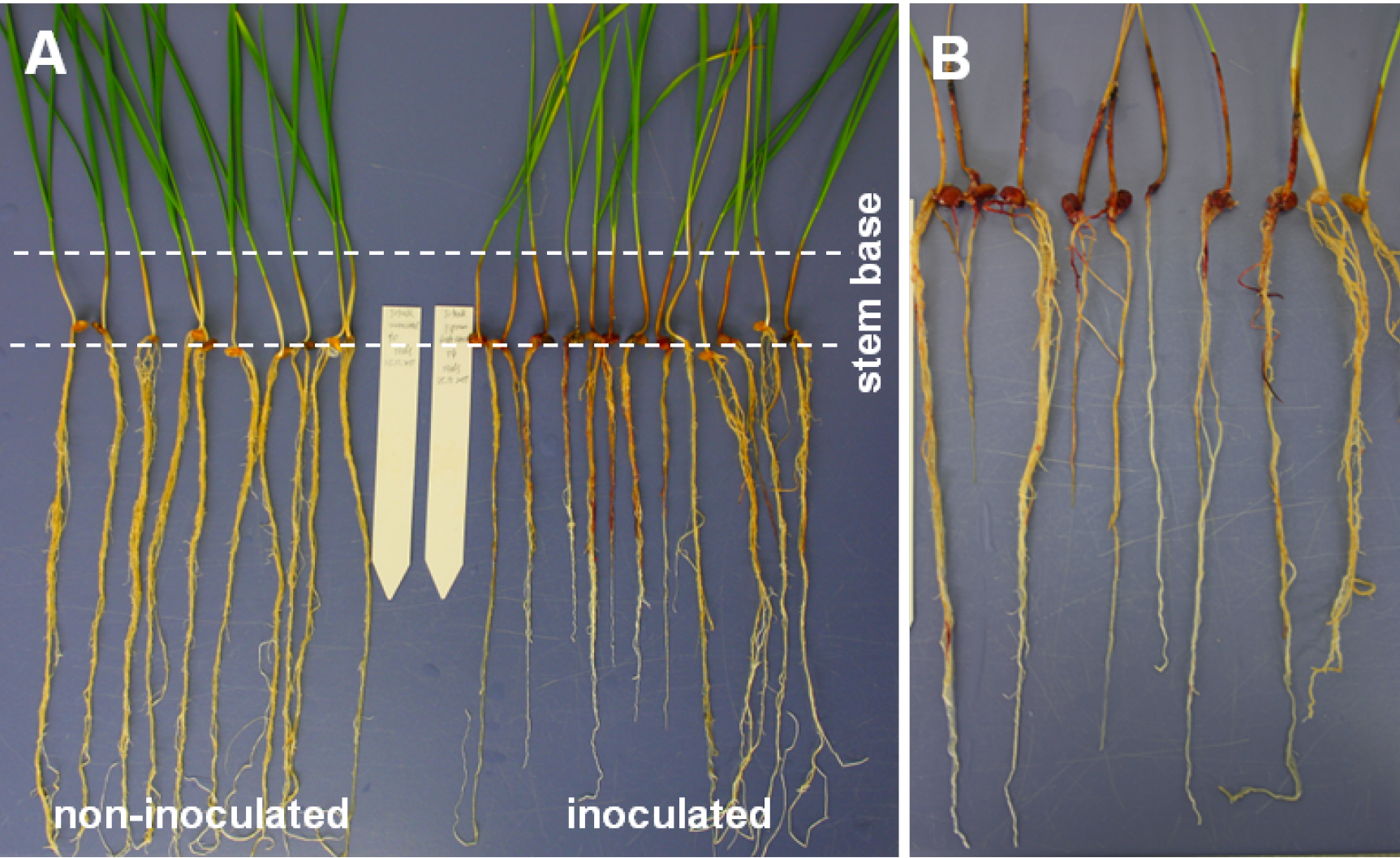
Figure 3. Wheat root phenotypes and necrosis symptoms. The example of FRR severity shows seedlings of the high-yielding cultivar Tobak. A. Seedlings at 14 days after root inoculation with Tween-20 (left) and F. graminearum (right). Inoculated seedlings show root and stem base necrosis as well as lowered root development. B. Close up of root and stem base symptoms visualised 21 days after root inoculation.
- Tissue sampling
- DNA preparation for reference-gene-based qPCR quantification
- Select and pool root tissues of five seedlings per genotype/treatment/timepoint to be used for fungal and root biomass quantification. Freeze root samples in liquid nitrogen and store them at -80 °C until further use. Extract gDNA from root tissues according to the protocol given by Doyle and Doyle (1990). Dilute gDNA of all samples to 50 ng/μl as work solution.
- Preparation of gDNA as a plant standard in the qPCR quantification of root biomass. Collect leave samples from non-inoculated seedlings or young plants. Dilute gDNA serially with sterile double-distilled water (100, 50, 25, 6.25, 1.5625, 0.39 ng wheat DNA/μl) and keep DNA at -20 °C until use.
- For the isolation of gDNA from F. graminearum mycelium, inoculate 100 ml potato dextrose broth (PDB) with 2 ml of 2 x 105 macroconidia suspension and incubate on a rotary shaker at 28 °C for 3 to 5 days (depending on the mycelium growth). Harvest the mycelium by filtration: pass the PDB through four layers of sterile cheese cloth. Collect the mycelium, grind in liquid nitrogen and store it at -80 °C by using a mortar and pestle. Isolate the gDNA by applying the CTAB-based method described by Brandfass and Karlovsky (2008).
- Preparation of gDNA as a Fusarium standard in the qPCR quantification of fungal biomass. Dilute gDNA serially with sterile double-distilled water (50, 10, 5, 1, 0.5, 0.1 ng F. graminearum DNA/µl) and keep the DNA at -20 °C until use.
- Select and pool root tissues of five seedlings per genotype/treatment/timepoint to be used for fungal and root biomass quantification. Freeze root samples in liquid nitrogen and store them at -80 °C until further use. Extract gDNA from root tissues according to the protocol given by Doyle and Doyle (1990). Dilute gDNA of all samples to 50 ng/μl as work solution.
- Reference-gene-based qPCR measurement of root and fungal biomass
- To amplify a F. graminearum specific fragment (Nicholson et al., 1998), use the primers Fg16N-F (ACAGATGACAAGATTCAGGCACA) and Fg16N-R (TTCTTTGACATCTGTTCAACCCA). To amplify wheat DNA (Ubiquitin gene, DQ086482) use the primers Ubi-F (CCCTGGAGGTGGAGTCATCTGA) and Ubi-R (GCGGCCATCCTCAAGCTGCTTA).
- Split PCR plate into two parts, each containing the wheat or fungal dilution series and eight root samples in three technical replicates. The amplification mix consists of 1 μl template DNA, 5 μl Roche FastStart Universal SYBR Green Master, 2 μl double-distilled water, and each 1 µl of forward and reverse primer (10 pmol/μl). Perform PCR with initial denaturation for 1.5 min at 95 °C; followed by 35 cycles with 30 sec at 94 °C, 45 sec at 64 °C, and 45 sec at 72 °C; and final elongation for 5 min at 72 °C.
- Analyse melting curves to ensure that only a single product was amplified. After amplifications, acquire the melting curves by heating the samples to 95 °C for 1 min, cooling to 55 °C for 1 min and then slowly increasing the temperature from 65 °C to 95 °C at the rate of 0.5 °C 10 sec-1, with continuous measurement of the fluorescence.
- Plot the CT values of each standard dilution series against the natural logarithm of the DNA concentration. Calculate the total amount of wheat DNA and fungal DNA with the gradient equation (f(x) = ax + b), according to the following formula (Livak and Schmittgen, 2001):
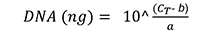
- Calculate the average amount of F. graminearum in wheat roots and the infection percentage of root samples (relative fungal biomass) by using the reference gene-based DNA quantification method and formula published by Brunner et al. (2009).
- Finally, calculate the relative fungal biomass as the ratio between fungal and wheat root DNA quantity:
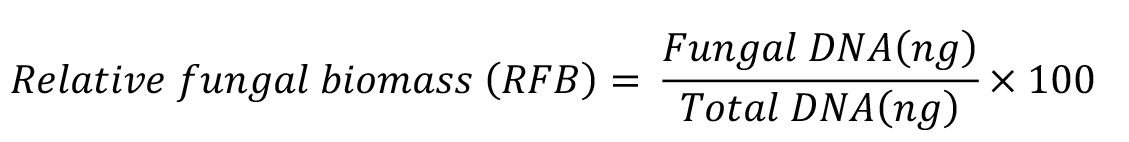
Where, total DNA is the sum of F. graminearum and wheat root DNA.
Note: Calculate the average infection percentage as the average of the assayed technical and biological replicates.
- To amplify a F. graminearum specific fragment (Nicholson et al., 1998), use the primers Fg16N-F (ACAGATGACAAGATTCAGGCACA) and Fg16N-R (TTCTTTGACATCTGTTCAACCCA). To amplify wheat DNA (Ubiquitin gene, DQ086482) use the primers Ubi-F (CCCTGGAGGTGGAGTCATCTGA) and Ubi-R (GCGGCCATCCTCAAGCTGCTTA).
- Fusarium root rot disease index (FDI)
The FDI incorporates all (or selected) severity parameter: relative fungal biomass; percentage change in root biomass, root length and shoot length; root and stem base symptom rating. Analogous to the area under the disease progress curve (AUDPC; Buerstmayr et al., 2000), the FDI can be used to rank genotypes for their overall performance under disease conditions (Wang et al., 2015) or as phenotypic parameter in genetic studies (Rutkoski et al., 2012).- Transfer the measured values of each severity parameter into dimensionless susceptibility index (SI) values to allow normalisation and direct comparison between different types of value. Calculate SI values using the equation:
SI = (XTx)/(Y)
Where,
X represents the measured value for each genotype and timepoint (Tx),
Y the corresponding overall mean calculated over all assayed genotypes and timepoints. - Calculate the respective FDI values for each genotype and timepoint as the sum of SI values.
Note: In time-course studies FDI values can be calculated for each sampling timepoint.
- Transfer the measured values of each severity parameter into dimensionless susceptibility index (SI) values to allow normalisation and direct comparison between different types of value. Calculate SI values using the equation:
Data analysis
Statistical analyses were performed by using the software SPSS 20 (IBM SPSS Statistics 20; IBM Corp., USA). The repeated measures ANOVA (rANOVA) was used to assess the time-course of FRR disease progression. In case the Mauchly’s test for sphericity violated the equality assumption, the Greenhouse-Geisser correction was applied to adjust the degrees of freedom appropriately. For multiple comparisons the Bonferroni adjustment (correction for Type I error) was applied. Two-way rANOVA was performed to test treatment and time effects on the traits root biomass, root length, and shoot length. Mixed rANOVA was performed to test for time effects (changes) in the disease progress in terms of fungal growth, reduction in root and shoot biomass of disease seedlings, and necrosis symptom development on roots/stem bases. Example tables for rANOVA results can be found in Wang et al., (2015).
Note: The mean profile plots (estimated marginal means) provided by the software SPSS 20 were applied to interpret significant time x treatment interactions. Examples for mean profile plots can be found in Wang et al. (2015). Tests of Within-Subjects Effects table provided by the software SPSS 20 gives three corrections for the significance of F-values: Sphericity assumed, Greenhouse-Geisser, Huynh-Feldt.
- Two-way rANOVA to test for significant treatment and treatment x time interactions in assessed severity traits [e.g., relative fungal biomass; root biomass and root length reduction; shoot length reduction]. This design includes the within-subjects factors ʻTimeʼ (independent factor with x sampling timepoints) and ʻTreatmentʼ (independent factor with FRR inoculated or non-inoculated control).
Note: The traits relative fungal biomass, root and stem base symptom index are not examinable in non-inoculated control plants and thus, cannot be test in two-way design. - The mixed rANOVA to test for significant time effects on the examined severity traits (dependent variables). This design includes the within-subjects factor ʻTimeʼ (as described for two-way rANOVA) and the between-subjects factor ʻGenotypeʼ (with x genotypes tested).
Note: Here, no comparison between treatments is included and thus, also those traits can be tested which are only examinable in inoculated plants, while traits examinable in both inoculated or non-inoculated plants have to be analysed as calculated relative reduction values (see step C2).
Notes
- Variation in the disease severity can result from differences in the aggressiveness of fungal isolate. A reduced aggressiveness (infection rate) can result from long time storage on agar media. Therefore, we recommend to prepare fresh macroconidia suspension from stock solution (stored at -80 °C) prior to infection experiments.
- We preferred the cultivation of inoculated plants in a growth chamber over the greenhouse. In the growth chamber infection and disease progression were more reliable and comparable between independent experiments. For investigations on host-pathogen interactions almost stable environmental conditions ensure that the pathogen is the most prevalent factor that influences plant development. Indeed, for investigations on adult plants a growth chamber may provide not enough space.
- If available marker genotype(s) with well-characterised response(s) to root infection should be added to experiments to ensure that root inoculation was successful. For instance, a markedly and early visible symptom development on the stem base allows rapid estimation of infection.
- The necrosis symptom assessment might be challenged by certain factors that should be taken into account. In the seedling stages, necrosis rating on stem bases can be hampered by an early natural (non-pathogenic) browning of control stem bases, a phenomenon displayed by certain genotypes. Root necrosis is occasionally not clearly recognizable, although fungal growth and disease severity measurements demonstrate susceptibility to root infection. Whether this phenomenon results from experimental issues or pathogen behaviour is currently unanswered. Generally, we observed that rather dry conditions seem to support formation of root necrosis by the pathogen. Moreover, we suggest that F. graminearum is capable of evolving a symptom-free (rather endophytic) life style under certain conditions (Wang et al., 2015; Mudge et al., 2006). In this context, the disease spread within upper stem internodes is generally symptom-free and can only be monitored by using qPCR quantification of fungal biomass (Procedure E) or diagnostic PCR marker for F. graminearum. In the adult plant stages, visual symptom assessment is, to our experiences, impossible due to the plant age-related natural browning of roots and stem bases.
Recipes
- 1.Synthetic nutrient deficient agar (SNA)
1 g KH2PO4
1 g KNO3
0.5 g MgSO4·7H2O
0.5 g KCl
0.2 g glucose
0.2 g sucrose
20 g agar
Dissolve in 1 L double-distilled water; then sterilise by autoclaving at 120 °C for 20 min
Acknowledgments
We would like to thank Prof. Herrmann Buerstmayr and Prof. Marc Lemmens (University of Natural Resources and Applied Life Sciences, Department for Agrobiotechnology, Vienna, Austria) for kindly providing the F. graminearum inoculum. This work was partially supported by China Scholarship Council. This protocol was adapted or modified from the study: Wang, Q., Buxa, S. V., Furch, A., Friedt, W. and Gottwald, S. (2015). Insights into Triticum aestivum seedling root rot caused by Fusarium graminearum. Mol Plant Microbe In 28(12): 1288-1303. The research paper can be downloaded via ResearchGate accounts of Qing Wang and Sven Gottwald.
References
- Beccari, G., Covarelli, L. and Nicholson, P. (2011). Infection processes and soft wheat response to root rot and crown rot caused by Fusarium culmorum. Plant Pathol 60:671-684.
- Brandfass, C. and Karlovsky, P. (2008). Upscaled CTAB-based DNA extraction and real-time PCR assays for Fusarium culmorum and F. graminearum DNA in plant material with reduced sampling error. Int J Mol Sci 9(11): 2306-2321.
- Brunner, K., Paris, M. P. K., Paolino, G., Burstmayr, H., Lemmens, M., Berthiller, F., Schuhmacher, R., Krska, R. and Mach, R. L. (2009). A reference-gene-based quantitative PCR method as a tool to determine Fusarium resistance in wheat. Anal Bioanal Chem 395(5): 1385-1394.
- Buerstmayr, H., Steiner, B., Lemmens, M. and Ruckenbauer, P. (2000). Resistance to Fusarium head blight in winter wheat: Heritability and trait associations. Crop Sci 40(4): 1012-1018.
- Doyle, J. J. and Doyle, J. L. (1990). A rapid total DNA preparation procedure for fresh plant tissue. Focus 12: 13-15.
- Leslie, J. F. and Summerell, B. A. (2006). The Fusarium laboratory manual. Blackwell Publishing 5: 388.
- Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods (25):402-408.
- Mudge, A. M., Dill-Macky, R., Dong, Y., Gardiner, D. M., White, R. G. and Manners, J. M. (2006). A role for the mycotoxin deoxynivalenol in stem colonisation during crown rot disease of wheat caused by Fusarium graminearum and Fusarium pseudograminearum. Physiol Mol Plant P 69: 73-85.
- Nicholson, P., Simpson, D. R., Weston, G., Rezanoor, H. N., Lees, A. K., Parry, D. W. and Joyce, D. (1998). Detection and quantification of Fusarium culmorum and Fusarium graminearum in cereals using PCR assays. Physiol Mol Plant P 53(1): 17-37.
- Rutkoski, J., Benson, J., Jia, Y., Brown-Guedira, G., Jannink, J. L. and Sorrells, M. (2012). Evaluation of genomic prediction methods for Fusarium head blight resistance in wheat. Plant Genome 5(2): 51-61.
- Trapero, C., Díez, C.M., Rallo, L., Barranco, D. and López-Escudero, F. J. (2013). Effective inoculation methods to screen for resistance to Verticillium wilt in olive. Sci Hortic 162: 252-259.
- Wang, Q., Buxa, S. V., Furch, A., Friedt, W. and Gottwald, S. (2015). Insights into Triticum aestivum seedling root rot caused by Fusarium graminearum. Mol Plant Microbe In 28(12): 1288-1303.
- Zadoks, J. C., Chang, T. T. and Konzak, C. F. (1974). A decimal code for the growth stages of cereals. Weed Res 14(6): 415-421.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wang, Q. and Gottwald, S. (2017). Wheat Root-dip Inoculation with Fusarium graminearum and Assessment of Root Rot Disease Severity. Bio-protocol 7(6): e2189. DOI: 10.21769/BioProtoc.2189.
Category
Microbiology > Microbe-host interactions > Fungus
Plant Science > Plant immunity > Disease bioassay
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.