- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protein Synthesis Rate Assessment by Fluorescence Recovery after Photobleaching (FRAP)

Published: Vol 7, Iss 5, Mar 5, 2017 DOI: 10.21769/BioProtoc.2156 Views: 12851
Reviewed by: Jyotiska ChaudhuriVarpu MarjomakiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2012

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Currently available biochemical methods cannot be applied to monitor protein synthesis in specific cells or tissues, in live specimens. Here, we describe a non-invasive method for monitoring protein synthesis in single cells or tissues with intrinsically different translation rates, in live Caenorhabditis elegans animals.
Keywords: Caenorhabditis elegansBackground
Proper regulation of protein synthesis is critical for cell homeostasis and growth. Deregulation of protein synthesis has been implicated in pathologies such as cancer and senescent decline (Bjornsti and Houghton, 2004; Syntichaki et al., 2007). Currently available biochemical methods for measuring general protein synthesis rate include metabolic labeling and polysomal profiling (Martin, 1998; Rennie et al., 1994). The applicability of these methodologies is limited due to poor intake and uncontrolled or unequal distribution of the label throughout the animal or tissue of interest. Also, these methods lack specificity and significant changes in specific cells or tissues of interest may be masked due to variability in intrinsic rates of translation of the bulk of the sample. In this protocol, we describe a method for monitoring protein synthesis rates in the nematode Caenorhabditis elegans, based on fluorescence recovery after photobleaching (FRAP). The experimental approach is based on the expression of fluorescent proteins, in cells and tissues of interest of transgenic animals. Fluorescence is then photobleached by irradiating cells, tissues or whole animals with a powerful light source. Recovery of fluorescence, indicative of new protein synthesis, is then monitored in cells or tissues of interest.
Materials and Reagents
- Greiner Petri dishes (60 x 15 mm) (Greiner Bio One, catalog number: 628161 )
- 35 mm plates (Corning, catalog number: 430165 )
- Microscope slides 75 x 25 x 1 mm (Marienfeld-Superior, catalog number: 10 006 12 )
- Microscope cover glass 18 x 18 mm (Marienfeld-Superior, catalog number: 01 010 30 )
- C. elegans strains (wild type [N2], ife-2[ok306], N2; Ex[pife-2GFP, pRF4], ife-2[ok306]; Ex[pife-2GFP, pRF4])
- Escherichia coli OP50 strain (obtained from the Caenorhabditis Genetics Center)
- Cycloheximide (Sigma-Aldrich, catalog number: C7698 )
Note: Cycloheximide has significant, toxic side effects. - Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: 7778-77-0 )
- Potassium phosphate dibasic (K2HPO4) (Sigma-Aldrich, catalog number: 7758-11-4 )
- Sodium chloride (NaCl) (EMD Millipore, catalog number: 106404 )
- Peptone (BD, Bacto, catalog number: 211677 )
- Streptomycin (Sigma-Aldrich, catalog number: S6501 )
- Agar (Sigma-Aldrich, catalog number: 05040 )
- Cholesterol stock solution (SERVA Electrophoresis, catalog number: 17101.01 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C5080 )
- Magnesium sulfate (MgSO4) (Sigma-Aldrich, catalog number: M7506 )
- Nystatin stock solution (Sigma-Aldrich, catalog number: N3503 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: 7558-79-4 )
- Phosphate buffer (1 M; sterile, see Recipes)
- Nematode growth medium (NGM) agar plates (see Recipes)
- M9 buffer (see Recipes)
Equipment
- Dissecting stereomicroscope (Nikon, model: SMZ645 )
- UV crosslinker (VilberLourmat, model: BIO-LINK – BLX-E365 )
- Epifluorescence microscope (ZEISS, model: Axioskop 2 Plus )
- Standard equipment for preparing agar plates (autoclave, Petri dishes, etc.) (Sambrook and Russell, 2001)
- Standard equipment for maintaining worms (platinum wire pick, incubators, etc.)
Note: For basic C. elegans culture, maintenance and manipulation techniques see refs (Epstein and Shakes, 1995; Lewis and Fleming, 1995; Hope, 1999; Strange, 2006). For information on C. elegans biology see refs (Wood, 1988; Epstein and Shakes, 1995; Riddle, 1997) and WormBook (http://www.wormbook.org/).
Software
- Camera control and imaging software (Carl Zeiss, Axio Vision 3.1 software)
- ImageJ image processing software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/) (Abràmoff et al., 2004)
- Microsoft Office 2011 Excel software package (Microsoft Corporation, Redmond, USA)
- Prism software package (GraphPad Software Inc., San Diego, USA)
Procedure
- Transgenic nematode generation and maintenance
To implement the protocol described below, start by generating appropriate transgenic C. elegans strains expressing the fluorescent proteins of choice in the cells or tissues of interest, under the control of promoters active for the cell or tissues investigated. Both transcriptional and translational fusions can be used with this method. Various methods are available for introducing recombinant DNA into nematodes. A widely exercised technique is the microinjection of DNA into the gonadal syncytium of gravid adult hermaphrodite animals (Rieckher et al., 2009).- Grow transgenic animals expressing the fluorophore in cells or tissues of interest on 60 mm plates seeded with OP50 (the E. coli strain used as C. elegans’ food source), at the standard temperature of 20 °C or other appropriate temperature (depends on genetic background and other experimental considerations such as the temperature sensitivity of animals examined).
- Unless an environmentally controlled chamber or room is available, the procedure of fluorescence photobleaching and recovery is performed at ambient temperature. Allow animals to equilibrate at this temperature for 2-3 h before photobleaching.
- Grow transgenic animals expressing the fluorophore in cells or tissues of interest on 60 mm plates seeded with OP50 (the E. coli strain used as C. elegans’ food source), at the standard temperature of 20 °C or other appropriate temperature (depends on genetic background and other experimental considerations such as the temperature sensitivity of animals examined).
- Sample preparation, photobleaching and recovery
- Sample preparation
The procedure can be performed either directly on a plate (step B1a) or on a coverslip (step B1b). When examining worms that express the fluorescent marker in individual cells, photographs of moving worms are hard to analyze. In this case follow step B1b, below. For worms expressing the fluorescent marker protein globally or in many tissues, the more convenient step B1a may be applicable. We generally avoid the use of anesthetics, especially the commonly used sodium azide, which blocks the mitochondrial respiratory chain, perturbs energy production and is likely to interfere with the fluorescence recovery process by hindering protein synthesis. To limit animal mobility (step B1a) we find particularly helpful the use of the dominant rol-6(su1006) allele as co-transformation marker (plasmid RF4), when constructing transgenic lines. This allele causes animals to roll instead of moving sinusoidally, which confines them in a relatively small area of the plate. - Transfer single worms to fresh 35 mm plates, seeded with OP50 bacteria. A small bacterial spot in the center of the plate will make localization of the worm easier, while focusing the sample.
- Spot a drop of 15 μl M9 buffer on a microscope slide and place the worm on the drop with the help of an eyelash glued on a pick. Add a cover slip on the top of the drop. The weight of the cover slip is sufficient to keep the worm immobile during the procedure, without damaging it.
Notes: - M9 buffer instead of water ensures a favourable osmotic environment for the worm. Animals should not be allowed to dry out during photobleaching. Supply fresh M9 in the form of 5 μl drops applied to the side of the cover slip during lengthy photobleaching sessions. (CRITICAL STEP)
- Exercise caution when removing the cover slip to recover the animal. Accidentally pressing on the cover slip will crush the worms. (CRITICAL STEP)
- Photograph single animals before photobleaching
Photograph single animals before photobleaching using a camera attached to the microscope (e.g., Axio Cam HR, Carl Zeiss). Images of fluorescent cells or tissues of interest are collected. Imaging parameters such as microscope and camera settings (lens and magnifier used, filters exposure time, resolution, etc.) should be documented. - Perform photobleaching
Use an epifluorescence, compound light microscope (e.g., Axioskop 2 Plus, Carl Zeiss) equipped with a high power light source (HBO 100; 100 Watt mercury arc lamp; Osram, Munich, Germany) and the appropriate excitation/emission filter sets to photobleach the animal. For the applications described here, 10 min of photobleaching reduce the initial emission intensity adequately (to within 30-50% of pre-bleach levels). The light intensity and the duration of the bleaching period are adjusted accordingly for the specific fluorophore, animal stage and cell or tissue under examination. Investigators should experimentally determine the appropriate duration of irradiation required to reach the extent of photobleaching, appropriate for different specimens (see Note 1).
Notes: - At least 20 individual animals should be processed for each experimental condition. The photobleaching period should be kept identical for all animals tested.
- CRITICAL STEP: Proper photobleaching conditions (light intensity, duration) should be set aiming to avoid injuring worms. The absolute level of fluorescence reduction by photobleaching is not important. We assess damage to worms by looking for apparent changes in behavior such as lethargy and movement defects or diminished responsiveness to touch, and for reduced fecundity in animals subjected to photobleaching. Animals showing signs of damage after photobleaching are excluded from further analysis.
- Fluorescence recovery
Photograph each animal immediately after photobleaching. Collect several images of cells or tissues of interest. Move animals to fresh OP50-seeded NGM plates. Recover animals photobleached on a microscope slide by adding 100 μl of M9 at the edges of the cover slip and sliding off the cover slip. These worms are also returned to an OP50-seeded NGM plate for recovery. Recovery timing starts at this point.
Note: All imaging parameters such as microscope and camera settings (lens and magnification used, filters exposure time, resolution, etc.) should be set as in step B2 above. (CRITICAL STEP) - Photographing animals at defined time points after fluorescence recovery
Follow fluorescence recovery by photographing animals at defined time points. We use 1 h intervals between successive photography sessions. A suitable time interval can be determined for each experimental application. Collect several images of cells or tissues of interest. As in step B4 above, it is critical that all imaging parameters (microscope and camera settings) are kept identical to those initially set in step B2 (see Note 2). - Prepare a stock solution of cycloheximide by diluting in water to a concentration of 10 mg/ml. Keep refrigerated. Add cycloheximide on top of OP50-seeded, 35 mm NGM plates to 500 μg/ml final concentration in the agar volume and allow plates to dry.
Notes: - The antibiotic cycloheximide, a potent and specific inhibitor of mRNA translation can be used to discriminate between the contribution of new protein synthesis and protein diffusion in overall fluorescence recovery after photobleaching.
- Cycloheximide has significant, toxic side effects. Kill bacteria on plates before adding cycloheximide by exposing bacterial lawns on NGM plates to UV radiation. Irradiate at 254 nm for 10 min at 100 mJ/cm2 in a UV crosslinker (Garigan et al., 2002; Gems and Riddle, 2000) (such as the Stratalinker 2400; Statagene, La Jolla, USA). (CAUTION)
- Transfer animals onto cycloheximide-containing plates and incubate for 2 h, at the growth temperature. Prepare animals for photobleaching as in step B1a or B1b. Return worms in cycloheximide-containing plates during fluorescence recovery (see Note 3).
- Sample preparation
Data analysis
- Image acquisition
Process images acquired in steps B2, B4 and B5 with ImageJ to determine the average and maximum pixel intensity for each image of fluorescent cell or tissue of interest in the collected photomicrographs. For each cell, tissue or animal, images should be acquired or converted to a pixel depth of 8 bit (256 shades of grey). Representative images of transgenic animals expressing pife-2GFP throughout somatic tissues in a wild type background or in IFE-2 deficient animals, in the presence or absence of cycloheximide, are shown in Figure 1. - Fluorescence intensity quantification
To analyze the area of interest manually, use the ‘freehand selection’ tool to enclose the fluorescent area. Select the ‘measurement’ command via the ‘analyze’ drop-down menu to perform pixel intensity analysis. On occasion (area continuity, high contrast ratios), selection of the fluorescent area can be done automatically. Select ‘adjust’ and then the ‘threshold’ command, within the ‘image’ drop-down menu of ImageJ. Adjust the threshold until the region of interest is marked. Within the ‘analyze’ drop-down menu, select the ‘analyze particles’ command. By selecting ‘outlines’ at the ‘show’ drop-down menu, check whether measurements correspond to the area of interest. Average and maximum pixel intensity values are collected for each transgenic line and grouped into ‘Pre-bleach’, ‘Bleach’ and ‘Recovery(n)’, where n is the time interval after photobleaching. - Statistical analysis
Statistical analysis of data is carried out using the Microsoft Office 2011 Excel software package (Microsoft Corporation, Redmond, USA) and the Prism software package (GraphPad Software Inc., San Diego, USA). The best-fit function that describes the recovery phase is generated by regression analysis (Figure 2). The slope of the best-fit lines provides quantification of the recovery rate. In the example shown, recovery is diminished in IFE-2-deficient animals or animals treated with cycloheximide.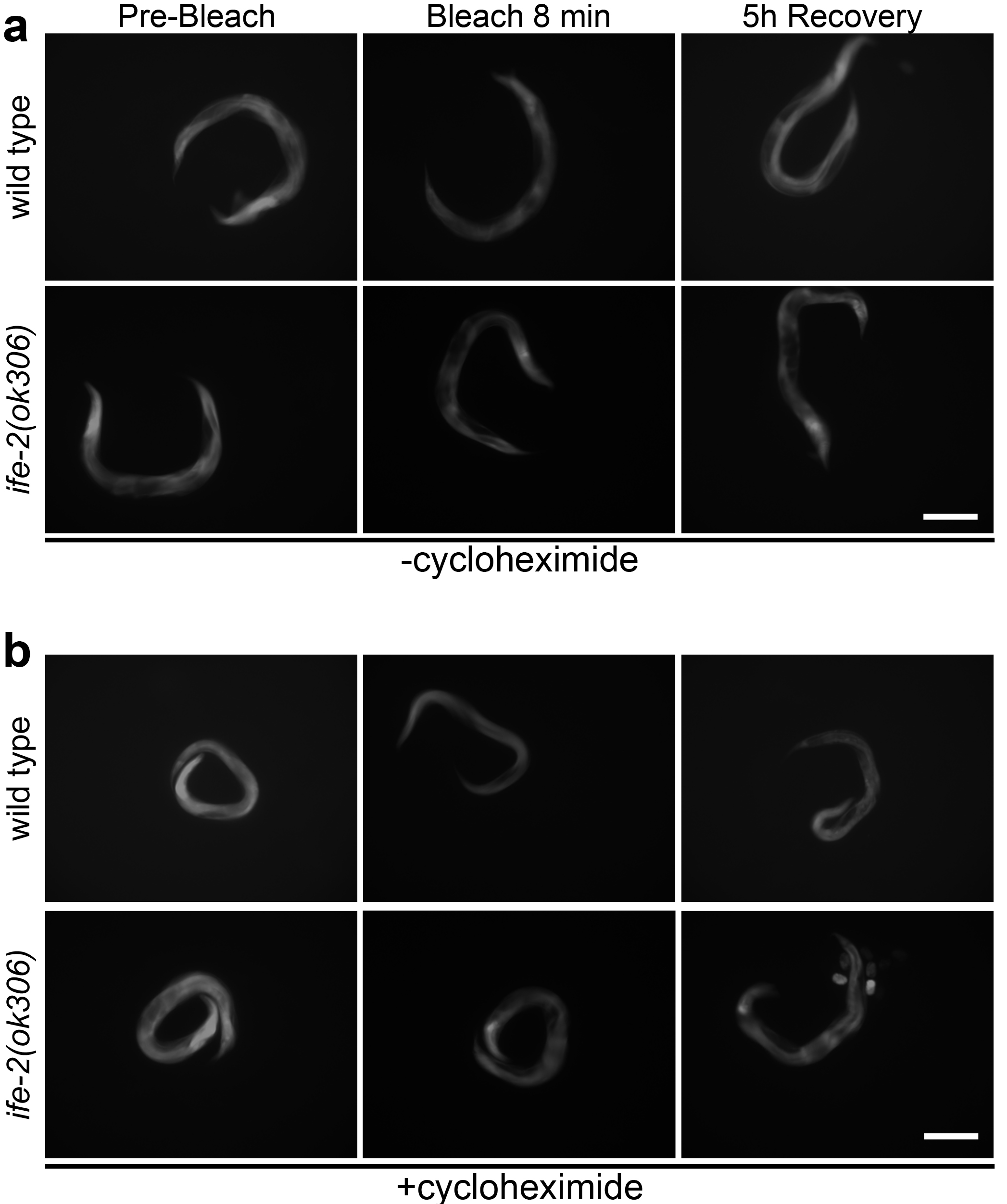
Figure 1. Photobleaching and recovery of fluorescence in vivo. Representative images of roller, transgenic animals expressing pife-2GFP throughout somatic tissues, in a wild type or IFE-2 deficient background before photobleaching, immediately following an 8 min whole-animal photobleaching session, and after a 5 h recovery period, in the absence (a) and presence (b) of cycloheximide (500 μg/ml). Scale bars = 100 μm.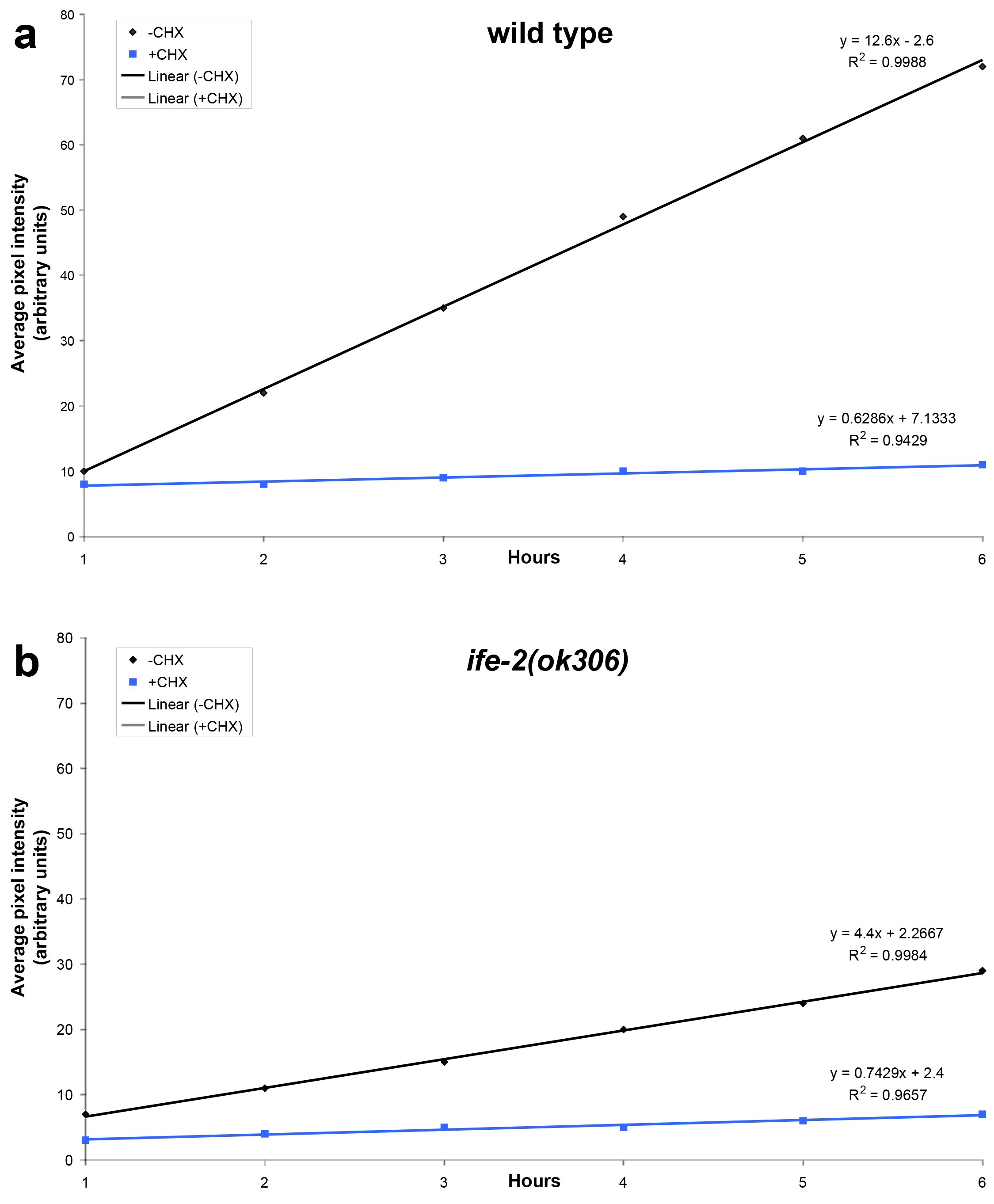
Figure 2. Regression analysis of fluorescence recovery in both wild type and IFE-2 deficient animals expressing pife-2GFP throughout somatic tissues. Best-fit lines are generated for average pixel intensity values obtained during the recovery phase for the indicated genetic backgrounds (a, wild type; b, ife-2[ok306]; black lines). The respective equations describing best-fit lines as well as R2 values for each line are also shown. Line slope corresponds to the first derivative of fluorescent change within a time unit (Δf/dt), which is a measure of the recovery rate. Cycloheximide treatment (CHX; 500 μg/ml) results in negligible recovery rate (blue lines).
Notes
- Inadequate photobleaching may be the result of insufficient duration of photobleaching and/or excitation light that is not concentrated on the tissue of interest. To overcome this problem, increase the duration of photobleaching, increase the light intensity or use an objective lens with higher magnification and numerical aperture.
- No significant recovery (step B5) may be an indication of specimen damage due to excessive photobleaching. It is also possible that the recovery kinetics are too slow for the chosen time interval. In addition, the promoter controlling the expression of the fluorophore may not be active during recovery. To address these issues, reduce photobleaching, allow longer recovery periods or switch promoter.
- Inadequate inhibition of recovery from photobleaching by cycloheximide may result from insufficient exposure to cycloheximide. Increase the pre-incubation time of the animals in the presence of cycloheximide.
Recipes
- Phosphate buffer (1 M)
- For 1 L, dissolve 102.2 g KH2PO4 and 57.06 g K2HPO4 in distilled water and fill up to 1 L. This is a 1 M solution, pH 6.0
- Autoclave at 121 °C for 15 min and store at room temperature
- Nematode growth medium (NGM) agar plates
- Mix 3 g NaCl, 2.5 g Bacto peptone, 0.2 g streptomycin, 17 g agar and add 900 ml distilled water. Autoclave at 121 °C for 15 min
- Let cool to 55-60 °C
- Add 1 ml cholesterol stock solution, 1 ml 1 M CaCl2, 1 ml 1 M MgSO4, 1 ml nystatin stock solution, 25 ml sterile 1 M phosphate buffer, pH 6.0, and distilled sterile water up to 1 L
- Pour about 8 ml medium per Petri dish and leave to solidify
- Store the plates at 4 °C until used
- M9 buffer
- Dissolve 3 g KH2PO4, 6 g Na2HPO4, 5 g NaCl in 1 L distilled water. Autoclave at 121 °C for 15 min
- Let cool and add 1 ml 1 M MgSO4 (sterile)
- Store M9 buffer at 4 °C
Acknowledgments
This work was funded by grants from the European Research Council (ERC), the European Commission 7th Framework Programme. The protocol has been adapted from Syntichaki et al. (2007), Nature 445, 922-926.
References
- Abràmoff, M. D., Magalhães, P. J. and Ram, S. J. (2004). Image processing with ImageJ. Biophotonics international 11(7): 36-42.
- Bjornsti, M. A. and Houghton, P. J. (2004). Lost in translation: dysregulation of cap-dependent translation and cancer. Cancer Cell 5(6): 519-523.
- Epstein, H. and Shakes, D. (1995). Caenorhabditis elegans: Modern biological analysis of an organism. Academic Press.
- Garigan, D., Hsu, A. L., Fraser, A. G., Kamath, R. S., Ahringer, J. and Kenyon, C. (2002). Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics 161(3): 1101-1112.
- Gems, D. and Riddle, D. L. (2000). Genetic, behavioral and environmental determinants of male longevity in Caenorhabditis elegans. Genetics 154(4): 1597-1610.
- Hope, I. A. (1999). C. elegans: a practical approach. OUP Oxford.
- Lewis, J. A. and Fleming, J. T. (1995). Basic culture methods. Methods Cell Biol 48: 3-29.
- Martin, R. (1998). Protein synthesis : methods and protocols. Humana.
- Rennie, M. J., Smith, K. and Watt, P. W. (1994). Measurement of human tissue protein synthesis: an optimal approach. Am J Physiol 266(3 Pt 1): E298-307.
- Riddle, D. L., Blumenthal, T., Meyer, B. J. and Priess, J. R. (1997). C. elegans II. Cold Spring Harbor Laboratory.
- Rieckher, M., Kourtis, N., Pasparaki, A. and Tavernarakis, N. (2009). Transgenesis in Caenorhabditis elegans. Methods Mol Biol 561: 21-39.
- Sambrook, J. and Russell, D. W. (2001). Molecular cloning: a laboratory manual 3rd edition. Cold Spring Harbor Laboratory.
- Strange, K. (2006). C. elegans: methods and applications. Humana.
- Syntichaki, P., Troulinaki, K. and Tavernarakis, N. (2007). eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 445(7130): 922-926.
- Wood, W. B. (1988). The nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kourtis, N. and Tavernarakis, N. (2017). Protein Synthesis Rate Assessment by Fluorescence Recovery after Photobleaching (FRAP). Bio-protocol 7(5): e2156. DOI: 10.21769/BioProtoc.2156.
Category
Developmental Biology > Cell signaling
Biochemistry > Protein > Synthesis
Biochemistry > Protein > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.