- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Yeast DNA Replication 2D Gel Protocol
Published: Vol 2, Iss 13, Jul 5, 2012 DOI: 10.21769/BioProtoc.213 Views: 15466
Original research article
The authors used this protocol in:

Sep 2008
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
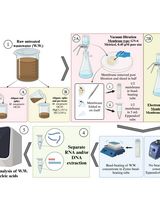
Two-dimensional agarose gel electrophoresis (2D gel) analysis is used extensively as a method to detect origins of replication. Here, I present a simplified method for the isolation of yeast genomic DNA for 2D gel analysis from a small number of yeast cells. This DNA isolation method is simpler and less time consuming than the traditional method that involves CsCl density gradient centrifugation. This method could be modified for 2D gel analysis in other organisms as well.
Materials and Reagents
- Qiagen genomic-tip 20/G ( QIAGEN, catalog number: 10223 , if more DNA needed, 100/G could be used to isolate DNA)
- Qiagen Buffer G2 ( QIAGEN, catalog number: 1014636 )
- Qiagen buffer QBT (QIAGEN, catalog number: 19054 )
- Qiagen buffer QC (QIAGEN, catalog number: 19055 )
- Qiagen buffer QF ( QIAGEN, catalog number: 19056 )
Tips: Buffer QBT, QC and QF could be made by yourself. - RNase A (10 mg/ml) ( QIAGEN, catalog number: 19103 )
- Proteinase K (20 mg/ml) (QIAGEN, catalog number: 19131 )
Tips: RNase A and Proteinase K could be ordered from any other company, they work fine too. - Sodium azide
- Isopropanol
- Tris-HCl
- EDTA
- Agarose
- KoAc
- Ethidium bromide
- Glycerol
- MgCl2
- Spermine
- Spermidine
- KOH
- GuHCl
- MOPS free acid
- Tween-20
- Triton X-100
- NaCl
- KB ladder
- 70% ethanol
- TE (see Recipes)
- Nuclear isolation buffer (NIB buffer) (see Recipes)
- Sodium azide solution (see Recipes)
- Qiagen Buffer QBT (see Recipes)
- Qiagen Buffer QC (see Recipes)
- Qiagen Buffer QF (see Recipes)
Equipment
- Acid-washed glass beads (425-600 μm diameter) (Sigma-Aldrich, catalog number: G8772-500G )
- BioRad sub-cell GT gel box or other Maxi horizontal electrophoresis units (big agarose gels are needed for both dimentions)
- 4 °C cold room
- Micro centrifuges
- Vortex mixer (VORTEX-GENIE)
- Microwave oven
- Hand-held UV lamp
- UV light box
- 2D gel apparatus tray (4 samples per apparatus -- Fischer self-circulating gel box)
- 50 ml polypropylene centrifuge tubes
- 2-ml polypropylene tube
- Pasteur pipette
- 12 x 75 mm polypropylene culture tube
Procedure
- Day 1: Solutions and preparation, as described in Materials and Reagents, and Recipes sections.
- Days 1-2: Yeast culture growth
- Pick a single colony or part of a single colony. Inoculate 5 ml cultures in the appropriate medium at the appropriate temperature.
- Enlarge the cell cultures after 1 overnight to OD600~0.1-0.2 in 50 ml medium.
- Grow until they reach log phase. Follow growth progress by monitoring OD600 over time: Harvest the cells when OD600 is between 0.6-1.0.
- Treat 50 ml cultures with 0.5 ml of 0.1 g/ml sodium azide (4.25 g NaAzide in 42.5 ml sterile H2O). Swirl well. Place on ice for 5-10 min.
- Harvest cells by centrifugation in 50 ml polypropylene centrifuge tubes, wash once with 20-30 ml of ice-cold distilled water and resuspend pellet in 0.8 ml ice-cold nuclear isolation buffer (NIB).
- Place cultures at -80 °C if doing the prep another day OR proceed to step 2 of the Genomic DNA purification protocol (below).
- Pick a single colony or part of a single colony. Inoculate 5 ml cultures in the appropriate medium at the appropriate temperature.
- Day 3: Genomic DNA Purification
- Thaw cultures from -80 °C freezer (frozen in NIB in the 2-ml polypropylene tube).
- An equal volume (800 μl) of acid-washed glass beads was added, and the cell suspension was vigorously shaken with a Vortex-Genie in the cold room at maximum speed for 1 min, and stood on ice for 1 min. This routine was repeated 12-20 times, until >90% of cells were broken (lysis monitored by phase contrast microscopy).
- Broken cells were removed with a Pasteur pipette to a 12 x 75 mm polypropylene culture tube, and the glass beads were rinsed twice with 0.8 ml ice-cold NIB and transferred to the same tube (it does not matter if some glass beads get transferred).
- Broken cells were separated into 2 of 1.5 ml eppendorf tubes and pelleted at 6,500 x g at 4 °C for 10 min and the pellet in each tube was dissolved in 1 ml Qiagen buffer G2 containing 20 μl RNase A (10 mg/ml).
- It is important to generate a homogeneous lysate by gently inverting the tube ~20 times (do not vortex).
- The lysate was incubated at 37 °C for 30 min, then 25 μl proteinase K (20 mg/ml) was added, and the lysate was incubated for 60 min at 42 °C.
- After incubation, the lysate was centrifuged at 6,500 x g at 4 °C for 10 min.
- A Qiagen column (20/G) was equilibrated with 2 ml Buffer QBT (allow the QIAGEN Genomic-tip empty by gravity flow).
- The supernatant from step 7 should be clear. Carefully transfer the supernatant to a 12 x 75 mm polypropylene culture tube. Pool the supernatants from the two tubes together.
Note: Take a 50 μl aliquot of the supernatant and save it for an analytic gel (aliquot 1). - Add an equal volume of Qiagen Buffer QBT to the supernatant and mix it by gently inverting the tube or Vortex it for 10 sec at full speed. Then load the sample to the equilibrated column and allowed to pass through by gravity flow.
Note: Take a 100 μl aliquot of the flow through and save it for an analytic gel (aliquot 2). - Wash the Column with 3 x 1 ml of Buffer QC.
Note: Take a 300 μl aliquot of the flow through and save it for an analytic gel (aliquot 3). - Elute the genomic DNA with 2 x 1 ml of Buffer QF. Use of Buffer QF pre-warmed to 50 °C will increase yields.
Note: Take a 50 μl aliquot of the flow through and save it for an analytic gel (aliquot 4). - Precipitate the DNA by adding 1.4 ml (0.7 volumes) room temperature isoproponal to the eluted DNA.
- The solution was well mixed and divided into two or three 1.5 ml Eppendorf tubes.
- Precipitate DNA by centrifugation in a microfuge at full speed for 15 min at 4 °C.
- The supernatants were discarded, 500 μl 70% ethanol (RT) was added to each tube.
- DNA was again pelleted for 2 min in a microfuge at full speed and most of the 70% ethanol was discarded. The tube was again centrifuged at full speed for 3-5 sec and all traces of 70% ethanol were removed.
- Air-dry for 5-10 min and resuspend DNA in 40 μl TE at RT for 1-2 h or at 4 °C overnight. If the DNA pellet is allowed to dry at this step, resuspension will be very difficult.
- At least three times during this time gently flick the tubes to promote resuspension of the genomic DNA. It is ok to SLOWLY pipette up and down using large orifice tips.
- Thaw cultures from -80 °C freezer (frozen in NIB in the 2-ml polypropylene tube).
- Day 4: Resuspension and pooling of genomic DNA
- After 24-30 h pool all the tubes for each sample (large orifice tips) and store at 4 °C for an additional 12+ h. Mix by slow pipetting up and down using large orifice tips.
- Do the analytic gel to determine yield, purity and length of DNA.
Precipitate each of aliquots with 0.7 volumes of isopropanal.
Rinse the pellets with 70% ethanol, drain well and resuspend in 20 μl of TE.
Use all 20 μl for analysis on a 0.5% agarose gel. Run the gel until the blue dye is near the bottom, and stain it in EtBr solution.
- After 24-30 h pool all the tubes for each sample (large orifice tips) and store at 4 °C for an additional 12+ h. Mix by slow pipetting up and down using large orifice tips.
- Day 6: Digestion of genomic DNA & starting the first dimension.
- Set up to digest ~10 μg of DNA per sample in a 600 μl reaction volume:
DNA + ddH2O should be equal a total volume of 150 μl, so add the appropriate volume of DNA to each tube to make 10 μg and bring the volume up to 150 μl.
Enzyme mix should be in a volume of 450 μl per sample. DNA is typically digested with 300-400 units of enzyme for 5-6 h at 37 °C. - Mix DNA with enzyme mixes by gently pipetting up and down using large orifice tips.
Note: Do not vortex your samples. - Digest DNA 5 h at 37 °C. Every hour gently mix DNA in tube by lightly flicking the tube and the bumping down and placing back at 37 °C.
- While DNA is digesting, make your 0.35% (w/v) agarose gel mix: 400 ml 1x TBE + 1.4 g Agarose in a 1 L glass bottle. Heat in the microwave until all agarose has gone into solution. Check for complete resuspension by swirling the bottle and looking at the bottom for agarose that is not fully in solution. Store at 55 °C for about 30-40 min to cool to a good pouring temperature. Pour the gel in the cold room. The gel tray should be 15 cm x 25 cm and should be leveled within the BioRad sub-cell GT gel box.1 gel allows for up to 8 samples. Once solidified, move gel to room temp. Set up for first dimension by adding the 1.6-1.8 L of 1x TBE needed to fill the gel box and then remove the comb.
- Confirm complete digestion by running an agarose gel of your digested DNA alongside undigested genomic DNA on a small 0.5% gel. Room for photo of gel(s) below.
- Precipitate digested DNA:
- Add 60 μl of 3 M KoAc (pH 5.5).
- Add 700 μl of 100% isopropanol.
- Mix gently by slowly inverting the tube several times.
- Add 60 μl of 3 M KoAc (pH 5.5).
- Spin down your precipitated DNA for 20 min at 14,000 rpm and 4 °C. Pour off the supernatent and wash the pellet 2x with 1 ml of 70% EtOH (5 min spins each time as above). Remove as much of the second wash as possible with a p200. Dry on the bench-top ~10 min. Resuspend with 16 μl of 1x TE (pH 8.0). Do not vortex. Resuspend by flicking the tube very gently and leaving on ice or at RT for ~1 h. After 1 h add 7 μl of 5x dyes, mix gently and bump down.
- Load KB ladder in the first lane. Skip two lanes and then load 20 μl of the DNA samples in every other lane.
- When done loading, start the gel. Run the gel 42-48 h at 22 V at RT.
- Set up for the next dimension by pre-chilling 2 L of 1x TBE for every 4 samples to be run. Place 4 L of buffer in the cold room. N.B. Add 60 μl of 10 mg/ml ethidium bromide to every 2 L of pre-chilled 1x TBE (0.3 µg/ml ethidium bromide final concentration) either now or right before using.
- Set up to digest ~10 μg of DNA per sample in a 600 μl reaction volume:
- Day 8: Starting the second dimension
- Make your 0.95% (w/v) agarose second dimension gel mixes: 500 ml of 1X TBE + 4.75 g of agarose + 15 μl of 10 mg/ml ethidium bromide (0.3 µg/ml final concentration). Mix in a 1 L glass bottle. Microwave on high until all agarose has gone into solution. Check by swirling the bottle and looking at the bottom. There should be no denser agarose visible. Store at 55 °C until ready to pour the second dimension.
- At the appropriate time stop the first dimension. Carefully soak the gel in a glass baking dish with 750 ml of 1x TBE with 0.3 µg/ml ethidium bromide to stain the DNA. This should take 30-40 min. The gel is very fragile, so be extra careful.
- Excise the KB ladder lane and photograph on the eagle-eye. Photograph a ruler alongside the ladder and overlay the two. Determine which 9-10 cm slice of the gel to run in the second dimension and excise by cutting between the lanes and with the right length. It is easiest to cut out the 9 cm slices first and then to cut between the lanes while on the UV light box. Try to cut with the UV setting on preparative (long wave) to avoid nicking. You can show the KB ladder and ruler in the space provided below:
- Transfer the slices to the 2D gel apparatus tray (4 samples per apparatus -- Fischer self-circulating gel box). Place the DNA so that the higher molecular weights are to the left. Set up the apparatus in the cold room and level it.
- Seal the edges of the gel and seal the slices in place with agarose. Pour the second dimension gel and make sure the gel slices do not move. Allow to solidify approximately 45 min to 1 h. Remove the gel dams and add the pre-chilled 2 L of 1x TBE and run the gel 18-30 h (depending on fragment size) at 130 V.
- You can monitor the progress in the second dimension with a hand-held UV lamp. Run the gel so that the smallest fragments on the arc of linear molecules are just reaching the bottom of the gel (lower right hand corner of the gel).
- Make your 0.95% (w/v) agarose second dimension gel mixes: 500 ml of 1X TBE + 4.75 g of agarose + 15 μl of 10 mg/ml ethidium bromide (0.3 µg/ml final concentration). Mix in a 1 L glass bottle. Microwave on high until all agarose has gone into solution. Check by swirling the bottle and looking at the bottom. There should be no denser agarose visible. Store at 55 °C until ready to pour the second dimension.
- Day 9: Stop second dimension and southern transfer
- Stop the gels. Cut the four gels apart in the middle, leaving you with two sets of two gels. Place in glass baking dishes and be sure to label the glass baking dishes with what is on each gel. Photograph the gels on the eagle eye (or UV light box).
- Paste gel photo(s) here.
- Go to the normal transfer process and southern blot steps.
- Stop the gels. Cut the four gels apart in the middle, leaving you with two sets of two gels. Place in glass baking dishes and be sure to label the glass baking dishes with what is on each gel. Photograph the gels on the eagle eye (or UV light box).
Recipes
- Nuclear isolation buffer (NIB buffer)
68 ml of 100% glycerol (17% v/v)
4.2 g of MOPS free acid (50 mM)
5.88 g of KoAc (150 mM)
0.8 ml of 1 M MgCl2 (2 mM)
20.8 mg of Spermine (150 µM)
200 μl of 1 M Spermidine (500 µM)
Add ddH2O to 375 ml, pH to 7.2 with KOH, add ddH2O to 400 ml.
Note: Do not autoclave. Store at 4 °C. - Sodium Azide (0.1 g/ml, 5 ml/500 ml culture)
42.5 ml ddH2O + 4.25 g Na-Azide
Vortex, store at 4 °C. - Qiagen Buffer G2 ( QIAGEN) (Bought) (pH 8.0)
800 mM GuHCl
30 mM EDTA
30 Mm Tris-HCl
5% Tween-20
0.5% Triton X-100 - Qiagen Buffer QBT (pH 7.0)
750 mM NaCl
50 mM MOPS
15% ethanol
0.15% Triton X-100 - Qiagen Buffer QC (pH 7.0)
1.0 M NaCl
50 mM MOPS
15% ethanol - Qiagen Buffer QF (pH 8.5)
1.25 M NaCl
50 mM Tris-HCl
15% ethanol - TE (pH 8.0)
10 mM Tris-HCl
1 mM EDTA
Acknowledgments
This protocol was adapted from Zou and Bi (2008) and Wu and Gilbert (1995).
References
- Wu, J. R. and Gilbert, D. M. (1995). Rapid DNA preparation for 2D gel analysis of replication intermediates. Nucleic Acids Res 23(19): 3997-3998.
- Zou, Y. and Bi, X. (2008). Positive roles of SAS2 in DNA replication and transcriptional silencing in yeast. Nucleic Acids Res 36(16): 5189-5200.
Article Information
Copyright
© 2012 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zou, Y. (2012). Yeast DNA Replication 2D Gel Protocol. Bio-protocol 2(13): e213. DOI: 10.21769/BioProtoc.213.
Category
Microbiology > Microbial genetics > DNA
Molecular Biology > DNA > DNA extraction
Molecular Biology > DNA > Electrophoresis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.