- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Histone H3 Cleavage Assay for Yeast and Chicken Liver H3 Protease
Published: Vol 7, Iss 1, Jan 5, 2017 DOI: 10.21769/BioProtoc.2085 Views: 9750
Reviewed by: Yanjie LiDamián Lobato-MárquezAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Histone proteins are subjected to a wide array of reversible and irreversible post-translational modifications (PTMs) (Bannister and Kouzarides, 2011; Azad and Tomar, 2014). The PTMs on histones are known to regulate chromatin structure and function. Histones are irreversibly modified by proteolytic clipping of their tail domains. The proteolytic clipping of histone tails is continuously attracting interest of researchers in the field of chromatin biology. We can recapitulate H3-clipping by performing in vitro H3 cleavage assay. Here, we are presenting the detailed protocol to perform in vitro H3 cleavage assay.
Keywords: Histone H3Background
Histone H3 clipping is the least understood mechanism of chromatin modification and regulation. It is expected that H3 clipping will permanently erase PTMs from the nucleosomes that might affect chromatin related events. Moreover, the fate of cleaved histones is still under investigation and it has been suggested that the cleaved histones might be recycled at specific regions of chromatin or they are targeted for degradation. There are various reports that describe in vivo clipping of histone H3 in different organisms, while in vitro assays for histone H3-specific clipping are limited. We need an efficient and robust in vitro assay for characterizing histone specific proteases. To this end, we present a protocol that can be used to examine the in vitro histone H3 clipping activity of yeast and chicken liver histone H3 proteases. We have optimized temperature and pH conditions for the assay. Under our optimized conditions, proteases were found to specifically cleave histone H3 out of all core histones. We have extensively used this protocol in our recent publications (Chauhan et al., 2016; Chauhan and Tomar, 2016; Azad and Tomar, 2016; Mandal et al., 2014; Mandal et al., 2013; Mandal et al., 2012). This protocol can be used to identify and characterize histone H3 specific proteases from different organisms ranging from yeast to mammals.
Materials and Reagents
- 1.5 ml centrifuge tubes (Tarsons)
- Dialysis tubing cellulose membrane (Sigma-Aldrich, catalog number: D9277 )
- Amicon ultra centrifugal filters (EMD Millipore, catalog number: UFC900396 )
- Cheese cloth
- Freshly extracted or frozen chicken brain tissue
- Saccharomyces cerevisiae yeast cells (BY4743 strain)
- Freshly extracted liver tissue from chicken
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- Sucrose (Sigma-Aldrich, catalog number: S7903 )
- Sodium chloride (NaCl) (EMD Millipore, catalog number: 567440 )
- Acetone (HiMedia Laboratories, catalog number: AS024 )
- Lyticase (Sigma-Aldrich, catalog number: L4025 )
- Protein A sepharose bead (GE Healthcare, catalog number: 17-0963-03 )
- Ammonium persulfate (Sigma-Aldrich, catalog number: A3678 )
- Ammonium sulfate [(NH4)2SO4] (Sigma-Aldrich, catalog number: A2939 )
- Coomassie Brilliant Blue R (Sigma-Aldrich, catalog number: B0149 )
- Potassium chloride (KCl) (EMD Millipore, catalog number: 104936 )
- Spermidine (Sigma-Aldrich, catalog number: S2626 )
- Spermine (Sigma-Aldrich, catalog number: 85590 )
- EDTA (Sigma-Aldrich, catalog number: EDS )
- EGTA (Sigma-Aldrich, catalog number: E3889 )
- β-mercaptoethanol (Sigma-Aldrich, catalog number: M3148 )
- Phenylmethanesulfonyl fluoride (PMSF) (Sigma-Aldrich, catalog number: 7626 )
- Disodium hydrogen phosphate (EMD Millipore, catalog number: 106586 )
- Sodium dihydrogen phosphate (EMD Millipore, catalog number: 106370 )
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- Glycerol (EMD Millipore, catalog number: 104093 )
- Protease inhibitor cocktail (PIC) (Sigma-Aldrich, catalog number: P2714 )
- Potassium acetate (Sigma-Aldrich, catalog number: P1190 )
- DTT (Sigma-Aldrich, catalog number: D9779 )
- Sorbitol (Sigma-Aldrich, catalog number: S6021 )
- Magnesium chloride hexahydrate (MgCl2) (Sigma-Aldrich, catalog number: 63064 )
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771 )
- Trizma base (Sigma-Aldrich, catalog number: T6066 )
- Hydroxyapatite resin (Sigma-Aldrich, catalog number: 289396 )
- Sulfuric acid (H2SO4) (EMD Millipore, catalog number: 112080 )
- Acrylamide (Sigma-Aldrich, catalog number: A8887 )
- GLUD1 antibody (Sigma-Aldrich, catalog number: SAB2100932-50UG )
Note: This Product has been discontinued. - Solution 1 (see Recipes)
- Hypotonic solution (see Recipes)
- Sodium phosphate buffer (1 M), pH 6.8 (see Recipes)
- HB buffer (see Recipes)
- HAP buffer (see Recipes)
- Potassium acetate solution (see Recipes)
- Prespheroplasting buffer (see Recipes)
- Spheroplasting buffer (see Recipes)
- Wash buffer (see Recipes)
- Lysis buffer (see Recipes)
- Protein A sepharose beads (50% slurry) (see Recipes)
- Yeast-protease resuspension buffer (see Recipes)
- Protease dialysis buffer (see Recipes)
- Reaction buffer
- Yeast protease reaction buffer (see Recipes)
- Chicken liver protease reaction buffer (see Recipes)
- 10% Triton X-100 solution (see Recipes)
- 6x SDS-PAGE loading dye (see Recipes)
- 10x SDS-PAGE running buffer (see Recipes)
Equipment
- Homogenizer (Pro Scientific, model: Bio-Gen PRO200 homogenizer )
- Potter-Elvehjem PTFE pestle and glass tube (Sigma-Aldrich, catalog number: P7734 )
- Centrifuge (Beckman Coulter, models: Avanti® J-E centrifuge-floor , Microfuge® 22R centrifuge )
- Sonicator (Boston Industries, model: Branson Digital Sonifier 250 & 102C Converter Ultrasonic Dismembrator )
Note: This product has been sold out. - Ultracentrifuge (Beckman Coulter, model: OptimaTM L-100K ultracentrifuge )
- Bio-Rad electrophoresis power supply (Bio-Rad Laboratories, model: PowerPacTM Basic Power Supply )
- Bio-Rad SDS-PAGE running apparatus (Bio-Rad Laboratories, model: Mini-PROTEAN tetra cell )
- Shaker (Eppendorf, model: New BrunswickTM Innova® 44 )
- Rotamer (Tarsons, catalog number: 3090 )
- Heating block, Thermocell Cooling and heating Block (Bio-Equip, model: HB-202 )
- FPLC cold cabinet (UniEquip, model: Unichromat 1500 )
- SuperoseTM 6, 10/300 GL chromatography column (GE Healthcare, catalog number: 17-5172-01 )
Procedure
- Preparation of core histones
- For protease assay, good quality of histones is required. We purified core histones from chicken brain tissue. To isolate histones from tissue requires preparation of nuclei as detailed below.
- Homogenize the chicken brain tissue (5 g) via a motor-driven homogenizer in solution 1 (50 ml) for 30 sec to make a 10% homogenate solution.
- Add 1.25 ml 10 % Triton X-100 drop-wise with constant shaking (swirling with hand) to the homogenate to a final concentration of 0.25%.
- Centrifuge the sample at 4 °C, 3,024 x g for 20 min and discard the supernatant to obtain nuclei pellet.
- Wash nuclei pellets (at 4 °C, 3,024 x g for 20 min) 3 times with 50 ml chilled solution 1 containing 0.34 M sucrose.
- Suspend the nuclei thoroughly into the 7 ml hypotonic buffer at a concentration of 1 mg DNA/ml. Check DNA concentration by measuring A280.
- Purify core histones from isolated nuclei by hydroxyapatite affinity purification (HAP) method as described below:
- Sonicate nuclei in 7 ml hypotonic solution to prepare soluble chromatin.
Note: We have observed that soluble chromatin preparation is fine when made as described above but this step can be improved. To improve soluble chromatin preparation, first dissolve nuclei in 7 ml HB buffer and apply 5 strokes by Potter-Elvehjem homogenizer. Centrifuge (27,216 x g, 4 °C for 5 min) the sample to collect nuclei. Repeat the same step (resuspension in HB buffer, homogenization, centrifugation and discard the supernatant) 6 times in total. - Further, dissolved nuclei in 7 ml HAP buffer containing 0.4 M NaCl and apply 5 strokes by hand homogenizer. Collect nuclei by centrifugation at 27,216 x g, 4 °C for 5 min. Repeat this step 2 times.
- Resuspend nuclei pellet in HAP buffer containing 0.6 M NaCl and sonicate (60% amplitude, 4 min-10 sec on, 10 sec at 4 °C) off it. These steps are adapted from Côté et al., 1995; Workman et al., 1991.
- Mix 5 g of hydroxyapatite resin (1.5 mg DNA/g hydroxyapatite) with 50 ml of 50 mM sodium phosphate buffer, pH 6.8. Keep it at room temperature for 10 min and centrifuge it (3,024 x g, 5 min). Discard the supernatant. Repeat it for two more times.
Note: Alternatively, equilibration of hydroxyapatite resin can be done with HAP buffer containing 0.6 M NaCl. - Mix soluble chromatin (1 mg/ml) with hydroxyapatite resin equilibrated in 50 mM sodium phosphate buffer, pH 6.8 (1.5 mg DNA/g hydroxyapatite).
- Add NaCl (5 M) to the final concentration of 0.6 M and incubate chromatin-HAP resin for half an hour at 4 °C with 10 rotations per minute on rotator.
- Wash chromatin mixed hydroxyapatite with 50 ml 50 mM phosphate buffer containing 0.6 M NaCl. Perform washing through centrifugation (3,024 x g, 5 min). Discard the supernatant. Repeat it 6 times.
- Elute total histones with 15 ml phosphate buffer containing 2.0 M NaCl. Perform the elution three times.
Note: Alternatively, we can elute histones with 15 ml HAP buffer containing 2.5 M NaCl. Perform the elution three times. - Desalt the eluted histones by dialysis against 1,000 ml of 10 mM Tris-Cl (pH 7.5) overnight at 4 °C with 4 changes.
Note: Alternatively, we can remove salt by spin filter and concentrate core histones, which can be used directly for assays. - Precipitate histones by addition of 3.5 volumes of chilled acetone. After adding acetone, keep the solution for precipitation at -20 °C for 1 h.
Collect the precipitated histones by centrifugation at 4 °C, 12,096 x g for 15 min and wash 2-3 times with chilled acetone.
Air-dry the pellet for 15 min and dissolve it in 500 μl 10 mM Tris-Cl (pH 7.5).
Check purity of core histones by running 18% SDS PAGE as shown in Figure1.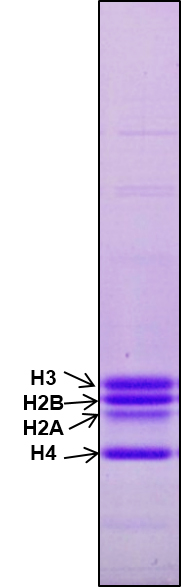
Figure 1. Purification of core histones. Core histones were purified from chicken brain tissue through hydroxyapatite resin and the purity was checked by running 18% SDS-PAGE. 2 μg core histones were loaded onto a lane.
- Purification of H3 protease
- Partial purification of H3-protease from Saccharomyces cerevisiae
- Make extract from sporulation-phase yeast cells (BY4743 strain) as it yields maximum H3-protease activity.
- Harvest 100 ml yeast cells growing in exponential phase (OD600-0.8) by centrifugation (2,300 x g, 5 min).
- Discard supernatant. Suspend yeast cell pellet in 2% (w/v) potassium acetate solution (50 ml).
- Allow the cells to grow further for 3 h at 30 °C with constant shaking (200 rpm). Under these condition, the yeast cells reach sporulation phase.
- Harvest yeast cells grown to sporulation-phase by centrifugation (2,300 x g, 5 min). Transfer the harvested cells into 1.5 ml centrifuge tubes. Wash the cell pellet with 2 ml of prespheroplasting buffer and discard the supernatant.
- Incubate the yeast cells in 0.5 ml spheroplasting buffer containing lyticase (250 U) at 37 °C for 15 min. Prepare the spheroplasts through lyticase enzymatic degradation of the yeast cell walls.
- Harvest the spheroplasts by centrifugation at low speed (3,300 x g) for 3 min at 4 °C.
- Discard the supernatant and wash the pellet once with 1 ml ice-cold wash buffer.
- Lyse the spheroplasts with 0.8 ml lysis buffer through incubation on ice for 15 min. Mix the cells by hand tapping at regular intervals.
- Centrifuge the cell lysates at 13,400 x g for 15 min at 4 °C to obtain clear lysates and transfer it into a new 1.5 ml centrifuge tube. Discard the pellet.
- To acquire H3-specific proteolytic activity, incubate the cell lysates with 100 µl of protein A sepharose beads (50% slurry) for 1 h at 4 °C with constant shaking on a rotator.
- Centrifuge (800 x g, 5 min, 4 °C) the bead mixed cell lysate and discard the supernatant.
- Similarly, wash the beads two times with 1 ml of lysis buffer (by reversing the microcentrifuge tube by hand, settle beads by centrifugation and throw the supernatant ) followed by washing twice with lysis buffer without Triton X-100.
- Resuspend the beads in yeast-protease resuspension buffer (100 µl) and use it for in vitro H3 cleavage assays.
- Partial purification of H3-protease from chicken liver
- Extract liver tissue from chicken and wash it with saline solution.
- Prepare the microsomes as described earlier (Grillo et al., 2002). Briefly, the method to purify microsomes from chicken liver tissue is described below.
- Homogenize 5 g chicken liver tissue via a motor-driven homogenizer in 50 ml of chilled solution 1 to make 10% homogenate and filter it through four layered cheese cloth.
- Remove nuclei and mitochondrial contaminations from homogenate subsequently by centrifugation at 5,000 x g, 4 °C for 10 min and 27,000 x g, 4 °C for 10 min respectively.
- Centrifuge the collected supernatant at 105,000 x g, 4 °C for 1 h to pellet down the microsomes.
- Resuspend the collected microsomes in 5 ml of 10 mM Tris-Cl, pH 8.0. Extract the resuspended microsomes with 0.2% Triton X-100 for 1 h at 4 °C by continuous rotation (rotator, 10 rpm).
- Centrifuge the extracted microsomes at 105,000 x g, 4 °C for 1 h. This time collect the supernatant and further precipitate it with 30% ammonium sulfate for 30 min at 4 °C.
- Collect the precipitates by centrifugation (27,216 x g, 4 °C, 30 min) and dialyze it over-night, against 2,000 ml dialysis buffer at 4 °C to remove ammonium sulfate.
- After dialysis, perform the centrifugation (27,216 x g, 4 °C, 15 min) to remove insoluble precipitates.
- To further purify H3 protease, perform SuperoseTM 6, 10/300 GL chromatography (Sample volume-500 μl, flow rate-0.3 ml/min, fraction volume-0.3 ml). Buffer for size exclusion chromatography is same as dialysis buffer. Collect positive fractions ranging from 39th to 51st as shown in Figure 2.
- Pool fractions (39th to 51st) together and use them directly (no need to concentrate these fractions for experiments) for in vitro histone H3 cleavage assay.

Figure 2. Western blot of liver microsomal extract with Glud1 antibody. Size exclusion chromatography fractions of chicken liver microsomal extract probed with Glud1 (Chicken liver H3 protease) antibody to confirm the presence of H3 protease in these fractions.
- Set up of in vitro histone H3 cleavage assay
- Protease source: yeast (BY4743 strain)
- Perform in vitro protease assay in reaction buffer (20 µl).
- Initiate assay by addition of 10 μl protein A sepharose bead-bound proteins into reaction mix containing 1-2 μg core histones.
- Incubate the reaction mix for 1 h at 37 °C, with constant shaking.
- Stop the in vitro cleavage reaction by addition of 3.33 μl 6x SDS-PAGE sample loading buffer into the reaction mixture (final concentration of SDS-loading dye in reaction mix: 1x).
- Protease source: chicken
- Perform the in vitro proteolytic assay in reaction buffer.
- Incubate 2 μg core histones with 0.25 μg of partially purified H3 protease extract from chicken liver tissue in 20 μl reaction volume.
- Incubate the reaction mix for 1 h at 37 °C.
- Stop the reaction by addition of 3.33 μl 6x SDS-PAGE sample loading buffer.
- Visualization of core histones after assay
- Stop the in vitro cleavage reaction by the addition of SDS-PAGE loading dye as mentioned earlier.
- Incubate the samples at 95 °C for 5 min.
- Examine the cleavage of histone H3 by running 18% SDS-PAGE as shown in Figure 3.
- Visualize the bands of core histones by staining with Coomassie blue staining.

Figure 3. In vitro histone H3 cleavage assay with core histones. A. In vitro activity assay with yeast protease. B. In vitro activity assay with chicken liver protease. Out of four core histones, only histone H3 get cleaved as protease is specific for histone H3. ‘-’ and ‘+’ indicate absence and presence of H3 protease respectively.
Note: The in vitro histone H3 cleavage assay works best when proteases are freshly prepared. The core histone should be stored in small aliquots at -80 °C. Repeated freeze-thawing of both histones and proteases should be avoided.
- Stop the in vitro cleavage reaction by the addition of SDS-PAGE loading dye as mentioned earlier.
Recipes
- Solution 1
0.34 M sucrose
15 mM Tris-Cl, pH 7.5
15 mM NaCl
60 mM KCl
0.5 mM spermidine
0.15 mM spermine
2 mM EDTA
0.5 mM EGTA
15 mM β-mercaptoethanol
0.2 mM PMSF (added before use) - Hypotonic solution
10 mM Tris-Cl, pH 7.5
15 mM β-mercaptoethanol
2 mM PMSF - Sodium phosphate buffer (1 M), pH 6.8 (100 ml)
49 ml of 1 M disodium hydrogen phosphate
51 ml of sodium dihydrogen phosphate - HB buffer
20 mM HEPES, pH 7.5
0.4M NaCl
1 mM EDTA
10% glycerol
0.1% NP-40
1 mM β-ME
PIC (Protease inhibitor cocktail) (added before use) - HAP buffer
50 mM sodium phosphate buffer
1 mM β-ME
PIC (Protease inhibitor cocktail) (added before use) - Potassium acetate solution
2 g potassium acetate dissolved in 100 ml water - Prespheroplasting buffer
100 mM Tris-Cl, pH 8.8
10 mM DTT - Spheroplasting buffer
50 mM Tris-Cl, pH 7.5
0.6 M sorbitol
10 mM DTT - Wash buffer
100 mM KCl
50 mM HEPES-KOH, pH 7.5
2.5 mM MgCl2
0.4 M sorbitol - Lysis buffer
20 mM Tris-Cl, pH 8.5
200 mM KCl
25 mM EDTA
1% (v/v) Triton X-100 - Protein A sepharose beads (50% slurry)
Take 500 µl protein A sepharose beads in 1.5 ml Eppendorf tube
Centrifuge it at 4 °C, 800 x g for 5 min
Discard supernatant
Add of 1 ml of lysis buffer to beads
Centrifuge it again and discard the supernatant
Repeat it two more time
Finally, resuspend the bead pellet in 500 µl of lysis buffer to obtain 50% slurry - Yeast-protease resuspension buffer
20 mM Tris-Cl, pH 8.5
200 mM KCl
25 mM EDTA - Protease dialysis buffer
25 mM Tris-Cl, pH 7.5
100 mM NaCl
10% glycerol
1 mM β-mercaptoethanol
0.2 mM EDTA - Reaction buffer
- Yeast protease reaction buffer
25 mM Tris-Cl, pH 7.5
150 mM NaCl
10% glycerol
2 mM β-mercaptoethanol
0.1 mM EDTA - Chicken liver protease reaction buffer
10 mM HEPES, pH 5.5
100 mM NaCl
1 mM β-mercaptoethanol
0.2 mM EDTA
10% glycerol - 10% Triton X-100 solution
10 ml of Triton X-100 from stock (100%)
90 ml water
Solution mixed well through rotation
10% solution
Stored at 4 °C - 6x SDS-PAGE loading dye
2.5 ml of 4x Tris-Cl, pH 6.8
3 ml glycerol
1 g SDS, 0.5 ml
1.2 mg bromophenol blue
Add water to make final volume to 10 ml - 10x SDS-PAGE running buffer
30.0 g of Trizma base
10.0 g of SDS
144.0 g of glycine
Dissolve in 1,000 ml of H2O
Dilute 10x buffer to 1x before use
Acknowledgments
This work was supported by the Department of Biotechnology (Government of India), Department of Science and Technology (Government of India) to RST. Short version of this protocol was published in Chauhan et al., 2016, Chauhan and Tomar, 2016; Azad and Tomar, 2016; Mandal et al., 2014; Mandal et al., 2013; Mandal et al., 2012. CSIR is acknowledged for providing fellowship support to Sakshi. Authors also acknowledge members of RST lab for their comments on this work.
References
- Azad, G. K. and Tomar, R. S. (2014). Proteolytic clipping of histone tails: the emerging role of histone proteases in regulation of various biological processes. Mol Biol Rep 41(5): 2717-2730.
- Azad, G. K. and Tomar, R. S. (2016). Partial purification of histone H3 proteolytic activity from the budding yeast Saccharomyces cerevisiae. Yeast 33(6): 217-226.
- Bannister, A. J. and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res 21(3): 381-395.
- Chauhan, S., Mandal, P. and Tomar, R. S. (2016). Biochemical analysis reveals the multifactorial mechanism of histone H3 clipping by chicken liver histone H3 protease. Biochemistry 55(38): 5464-5482.
- Chauhan, S. and Tomar, R. S. (2016). Efficient expression and purification of biologically active human cystatin proteins. Protein Expr Purif 118: 10-17.
- Côté, J., Utley, R. T., and Workman, J. L. (1995). Basic analysis of transcription factor binding to nucleosomes. Methods Mol Genet 6:108-128.
- Grillo, C., Coppari, S., Turano, C. and Altieri, F. (2002). The DNA-binding activity of protein disulfide isomerase ERp57 is associated with the a(') domain. Biochem Biophys Res Commun 295(1): 67-73.
- Mandal, P., Azad, G. K. and Tomar, R. S. (2012). Identification of a novel histone H3 specific protease activity in nuclei of chicken liver. Biochem Biophys Res Commun 421(2): 261-267.
- Mandal, P., Chauhan, S. and Tomar, R. S. (2014). H3 clipping activity of glutamate dehydrogenase is regulated by stefin B and chromatin structure. FEBS J 281(23): 5292-5308.
- Mandal, P., Verma, N., Chauhan, S. and Tomar, R. S. (2013). Unexpected histone H3 tail-clipping activity of glutamate dehydrogenase. J Biol Chem 288(26): 18743-18757.
- Workman, J. L., Taylor, I. C., Kingston, R. E. and Roeder, R. G. (1991). Control of class II gene transcription during in vitro nucleosome assembly. Methods Cell Biol 35: 419-447.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chauhan, S., Azad, G. K. and Tomar, R. S. (2017). In vitro Histone H3 Cleavage Assay for Yeast and Chicken Liver H3 Protease. Bio-protocol 7(1): e2085. DOI: 10.21769/BioProtoc.2085.
Category
Cancer Biology > Cancer biochemistry > Protein
Biochemistry > Protein > Activity
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.