- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation, Culture, and Staining of Single Myofibers


Published: Vol 6, Iss 19, Oct 5, 2016 DOI: 10.21769/BioProtoc.1942 Views: 16363
Reviewed by: Jia LiIrit AdiniAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Adult skeletal muscle regeneration is orchestrated by a specialized population of adult stem cells called satellite cells, which are localized between the basal lamina and the plasma membrane of myofibers. The process of satellite cell-activation, proliferation, and subsequent differentiation that occurs during muscle regeneration can be recapitulated ex vivo by isolation of single myofibers from skeletal muscles and culturing them under suspension conditions. Here, we describe an improved protocol to evaluate ex vivo satellite cells activation through isolation of single myofibers from extensor digitorum longus (EDL) muscle of mice and culturing and staining of myofiber-associated satellite cells with the markers of self-renewal, proliferation, and differentiation.
Keywords: Single myofiber culturesBackground
Although skeletal muscle is a fully differentiated post-mitotic tissue, it maintains intrinsic capability to regenerate in response to both genetic and acquired forms of muscle fiber damage (Le Grand and Rudnicki, 2007). Muscle regeneration in adults is mediated by a population of stem cells known as satellite cells which reside between the basal lamina and sarcolemma of myofibers in a mitotically quiescent state (Le Grand and Rudnicki, 2007). In response to muscle trauma, satellite cells become activated and proliferate to produce myoblasts that fuse with pre-existing fibers and with one another to repair or produce new myofibers. A small portion of satellite cells does not differentiate but instead reenters quiescence to maintain the stem cell pool. Satellite cells of all mammalian species express paired-box (Pax) transcription factor Pax7 which is also used as a critical marker to determine the fate of satellite cells in association with other myogenic factors such as MyoD (Le Grand and Rudnicki, 2007; Kuang et al., 2006; Kuang and Rudnicki, 2008).
The in vivo process of muscle regeneration with respect to satellite cell activation, proliferation, and differentiation can be partly recapitulated through suspension culture of myofiber explants (Rosenblatt et al., 1995; Shefer and Yablonka-Reuveni, 2005). The process of myofiber isolation which involves digestion of muscle tissue with matrix-degrading enzymes (e.g., collagenases) and mechanical shearing causes minor trauma which leads to the activation of satellite cells on myofibers. Immediately upon isolation, each myofiber is associated with a fixed number (Pax7+/MyoD-) of satellite cells resting in quiescence. At around 24 h in culture, satellite cells undergo their first round of cell division, through upregulating MyoD (Pax7+/MyoD+) and proliferate to form cell aggregates by 72 h. Satellite cells on cultured myofiber explants can either terminally differentiate (Pax7-/MyoD+) or self-renew (Pax7+/MyoD-) and return to quiescence (Le Grand and Rudnicki, 2007). The myofiber culture system has served as an excellent platform to study the fundamental properties of muscle stem cells and the effects of various genetic manipulations on the basic properties of satellite cells. One of the major advantages of isolated single myofiber cultures is that the physical interaction between the myofiber and satellite cells is preserved in the sense that satellite cells are still maintained beneath the basal lamina (Bischoff, 1986). Furthermore, suspension cultures of myofibers are routinely used to study the effects of various pharmacological compounds and overexpression or knockdown of specific proteins on self-renewal, proliferation, or differentiation of satellite cells (Shefer and Yablonka-Reuveni, 2005; Anderson et al., 2012; Keire et al., 2013). They also provide a useful tool to study the motility of satellite cells on myofibers through live imaging (Siegel et al., 2009). In addition, a few investigators have used isolated single myofibers explants to prepare pure myoblast cultures in which the cells are mostly derived from the myofiber-associated satellite cells.
The isolation of single myofibers from flexor digitorum brevis (FDB) was first described by Bischoff (1986). This protocol was modified later by Rosenblatt et al. to allow handling of single myofibers after collagenase digestion (Rosenblatt et al., 1995). Since then several modifications have been proposed which improved the yield and the handling of the isolated myofibers (Shefer and Yablonka-Reuveni, 2005; Anderson et al., 2012; Verma and Asakura, 2011; Pasut, 2013). However, most of the published protocols still do not produce a sufficient number of myofibers in a single preparation primarily because of under or over digestion with collagenase and the loss of myofibers during picking, sub-culturing, or staining. The amount of time to digest a muscle from rodents may vary with genetic background and healthy versus diseased muscle. In our laboratory, we are now using a six-well tissue culture plate in which the digestion and physical separation of individual myofibers can be visually monitored under a phase contrast microscope. Furthermore, we have developed an improved protocol to stain the myofiber-associated satellite cells for various markers (Hindi et al., 2012; Hindi and Kumar, 2016; Ogura et al., 2013). In this article, we provide the detailed protocol for the isolation, culturing, and staining of myofiber-associated satellite cells for Pax7 and MyoD protein. The same protocol can be adapted for staining of other proteins in satellite cells of myofiber explant.
Materials and Reagents
- Disposable borosilicate glass Pasteur pipets (Thermo Fisher Scientific, Fisher Scientific, catalog number: 13-678-20B )
- Sterilization pouches (Thermo Fisher Scientific, Fisher Scientific, catalog number: 01-812-51 )
- 6-well plates (Corning, Falcon®, catalog number: 353046 )
- 0.22 µm filter (EMD Millipore, catalog number: SLGP033RS )
- 0.45 µm filter (EMD Millipore, catalog number: SLHV033RS )
- 24-well plates (Corning, Falcon®, catalog number: 353047 )
- Adult mice (Mus musculus; ≥ 6-week old)
- Horse serum (Thermo Fisher Scientific, GibcoTM, catalog number: 26050-088 )
- Dulbecco’s modified Eagle’s medium (DMEM) high glucose, pyruvate (Thermo Fisher Scientific, GibcoTM, catalog number: 11995-065 )
- N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES) (1 M) (Thermo Fisher Scientific, GibcoTM, catalog number: 15630-080 )
- Penicillin-streptomycin (Pen/Strep) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140-122 )
- Collagenase II (Worthington Biochemical Corporation, catalog number: LS004176 )
- Ultra-pureTM water (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10977-015 )
- Phosphate buffered saline (PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10010-023 )
- 2, 2, 2-tribromoethanol (Avertin) (Sigma-Aldrich, catalog number: T48402 )
- 100% ethanol (Thermo Fisher Scientific, Fisher Scientific, catalog number: 2701 )
- Tris base (Thermo Fisher Scientific, Fisher Scientific, catalog number: BP152-5 )
- Recombinant human fibroblast growth factor-basic (bFGF) (PeproTech, catalog number: 100-18B )
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2153 )
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10437-028 )
- Chicken embryo extract (CEE) (Antibody Production Services, catalog number: MD-004E )
- Paraformaldehyde (PFA) (Sigma-Aldrich, catalog number: P6148 )
- 100% Triton X-100 (Thermo Fisher Scientific, Fisher Scientific, catalog number: BP151-500 )
- Glycine (Thermo Fisher Scientific, Fisher Scientific, catalog number: BP381-5 )
- 5% (w/v) sodium azide (Thermo Fisher Scientific, Fisher Scientific, catalog number: 71448-16 )
- Primary antibody anti-MyoD (rabbit) (Santa Cruz Biotechnology, catalog number: sc-304 )
- Primary antibody anti-Pax7 (mouse) (Developmental Studies Hybridoma Bank, catalog number: PAX7 )
- Secondary antibody goat anti-rabbit Alexa Fluor® 488 conjugate (Thermo Fisher Scientific, catalog number: A-11034 )
- Secondary antibody goat anti-mouse Alexa Fluor® 568 conjugate (Thermo Fisher Scientific, catalog number: A-11004 )
- 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma-Aldrich, catalog number: D8417 )
- Collagenase II (see Recipes)
- Digestion medium (see Recipes)
- Washing solution (see Recipes)
- 70% ethanol (see Recipes)
- Basic fibroblast growth factor (bFGF) (see Recipes)
- Chicken embryo extract (CEE) (see Recipes)
- Myofiber growth medium (MfGM) (see Recipes)
- 4% paraformaldehyde (PFA) (see Recipes)
- 10% Triton X-100 (see Recipes)
- Quenching solution (see Recipes)
- Blocking solution (see Recipes)
Equipment
- Water bath (Thermo Fisher Scientific, Fisher Scientific, catalog number: 15-462-15Q )
- Microscope (Nikon Instruments, model: TE2000 )
- CO2 incubator (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3578 )
Procedure
- Before isolation of myofibers
- Borosilicate glass Pasteur pipets ends are too sharp for myofibers. Flame polish them to smooth the end portion and then autoclave (see Note 1).
- Autoclave one set of tools containing one small pair of scissors and forceps in a sterilization pouch for extensor digitorum longus (EDL) muscle removal from mouse hind limbs.
- Coat 6-well plates with around 1 ml of horse serum (horse serum is used to study satellite cells activation as it keeps myofibers suspended). Let plates sit for 1 min, then remove excess (that you can put back in the original bottle to re-use multiple times, if kept sterile). Avoid bubbles. Leave plates open to dry (drying requires ~20 min). Close plates once they are dried and leave under hood at room temperature till ready to use. These plates will be used for isolation and culturing of myofibers following digestion of EDL muscle with collagenase.
- Following the recipe, prepare base of digestion medium (see Note 2).
- Following the recipe, prepare collagenase II (see Note 3).
- Following the recipe, prepare washing solution. To wash EDL muscle before digestion, prepare a 6-well plate with 3 ml per well of washing solution. Each well is specific for one pair of EDL muscles (right and left). There will be 3 washes per pair of EDL.
- Borosilicate glass Pasteur pipets ends are too sharp for myofibers. Flame polish them to smooth the end portion and then autoclave (see Note 1).
- Muscle removal and washing
- Euthanize mouse by administrating an IP injection of Avertin (2, 2, 2-tribromoethanol) at a dose of 250 mg/kg, followed by cervical dislocation.
- Spray 70% ethanol on the whole body and pin the mouse face up.
- Shave hair around hind limb area and gently remove the skin using autoclaved scissors and forceps (see Note 4).
- Remove carefully fascia surrounding right tibialis anterior (TA) muscle (Video 1).
 Video 1. Step-wise procedure for the isolation of EDL muscle from mouse
Video 1. Step-wise procedure for the isolation of EDL muscle from mouse - Remove carefully TA muscle without damaging underneath EDL muscle (see Note 5). The first muscle removed is TA muscle which can be discarded (Video 1).
- Remove carefully EDL muscle (Figure 1). Second muscle removed in Video 1 is EDL muscle. Avoid stretching of muscle during and after isolation (see Notes 5-7).
- Put EDL muscle in the well containing washing solution (first wash).
- Perform isolation of the other side EDL (try to do this step within next 5 min).
- Put EDL muscle in the same well as the other EDL muscle.
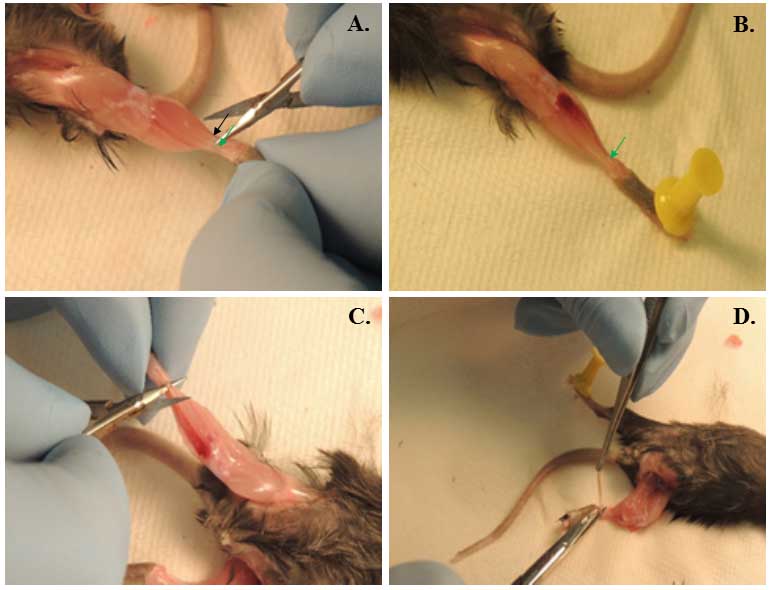
Figure 1. Isolation of EDL muscle from mice. A. Picture presented here depicts tendon for TA (black arrow) and EDL (green arrow) muscle; B. Mouse hind limb after removal of TA muscle. Arrow points to the tendon of EDL muscle; C. Separating EDL muscle from tendon to tendon; D. Isolation of EDL muscle by cutting distal tendons. - Spray the 6-well plate with 70% ethanol before putting it under the biosafety cabinet.
- Perform second and third washes (30 sec each), by transferring EDL muscle to the next well containing washing solution.
- Euthanize mouse by administrating an IP injection of Avertin (2, 2, 2-tribromoethanol) at a dose of 250 mg/kg, followed by cervical dislocation.
- Muscle digestion
- Following the recipe, complete the preparation of digestion medium by adding 400 U/ml of collagenase II to the base of digestion medium. Filter the digestion medium with a 0.22 µm filter.
- Take a new 6-well plate and put 5 ml of digestion medium per well (for two EDL muscles).
- Take autoclaved forceps and remove EDL muscles from the 6-well plate used for washing and put them in the 6-well plate containing digestion medium.
- Put the 6-well plate containing EDL muscles for 1.5 h to 2 h in a CO2 incubator set at 37 °C and 5% of CO2. After the first hour of digestion, check the condition of digesting EDL muscles by putting the 6-well plate under an inverted microscope and visualizing if single myofibers are released from the EDL muscle (see Note 8). Using sterile Pasture pipette and digestion medium, gently triturate EDL muscle to accelerate dissociation of single myofibers. Avoid over-digestion in collagenase (Figure 2). Excessive digestion generally results in the isolation of hyper-contracted myofibers (see Note 9).
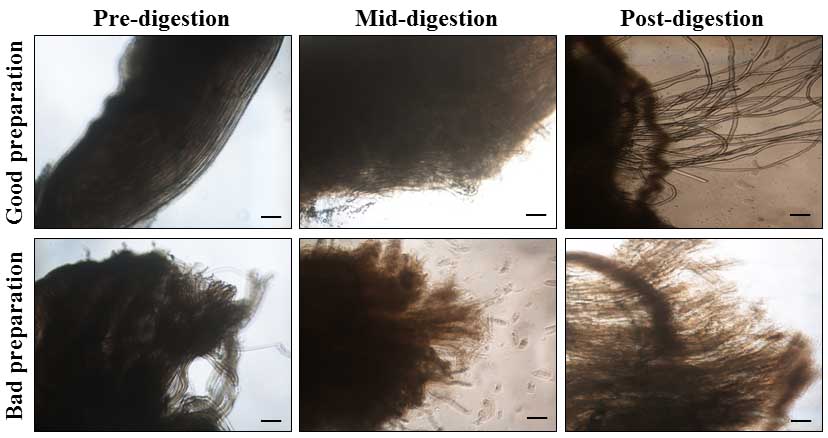
Figure 2. Digestion of EDL muscle using collagenase. Representative phase contrast microscopy images presented here show the morphology of good quality EDL muscle at different stages of collagen digestion (top panel). Top-left image shows tightly bundled fibers of EDL muscle prior to digestion. Top-middle image demonstrates partial dissociation of connective tissue at about 45 min of start of collagenase treatment. Top-right image depicts complete digestion and release of myofibers from EDL muscle. Lower panel images demonstrate corresponding phases of muscle digestion in an improperly isolated EDL muscle. Scale bar = 100 µm.
- Following the recipe, complete the preparation of digestion medium by adding 400 U/ml of collagenase II to the base of digestion medium. Filter the digestion medium with a 0.22 µm filter.
- Isolation and culturing of myofibers
- Following the recipe, prepare myofiber growth medium (MfGM).
- Coat autoclaved Pasteur pipets with horse serum (horse serum prevents myofibers from sticking to glass pipet).
- Take a new 6-well plate and put 2 ml of MfGM per well (for neutralization of collagenase activity following EDL muscle digestion).
- Remove 6-well plate containing EDL muscle from the incubator. Observe EDL muscle under the microscope to ensure normal structure and shape of single myofibers.
- Transfer digested EDL muscles (using autoclaved-flame polished Pasteur pipet) to a new 6-well plate containing 2 ml of MfGM in three wells (see Note 10).
- Transfer digested EDL muscles from one well to the next in a serial manner with autoclaved Pasteur pipet. Allow digested EDL’s to set in each of the 3 wells for 30 sec. This will neutralize the collagenase enzymatic activity and stop the digestion process.
- Take 6-well plates coated previously with horse serum and put 3 ml of MfGM per well (for culturing of myofibers).
- Transfer neutralized EDL muscles in the coated 6-well plate containing MfGM (i.e., well #1). Using autoclaved coated Pasture pipet, gently flush EDL muscle and myofiber bundles with MfGM. Do not vigorously triturate the muscle as it can cause damage to myofibers (see Note 11).
- Transfer the EDL muscles from well #1 to well #2. Flush gently EDL with autoclaved coated Pasteur pipet.
- Move the EDL muscles from well #2 to well #3 and repeat the same process until well #6. The goal is to have myofibers from EDL muscles in each well. Representative images of isolated single myofibers are presented in Figure 3.

Figure 3. Images of normal and hypercontracted isolated single myofiber cultures. Left image represents isolated single myofibers following a successful isolation. Fibers are mostly straight and translucent and their surface is clear of any shears or tears. Right image shows fiber morphology following an inadequate preparation. Red arrows point to hypercontracted fibers while blue arrows point to fragmented fibers. Scale bar = 100 µm. - Observe EDL myofibers under the microscope for their shape and structure (see Note 12).
- Put the 6-well plate containing EDL myofibers in a CO2 incubator set at 37 °C and 5% of CO2. If analysis of satellite cells is desired in the quiescent stage, fix myofibers immediately and proceed to staining. For studying activation of satellite cells, culture myofibers for desired time points. Normally, large satellite cell clusters are observed at 72 h post isolation.
- Following the recipe, prepare myofiber growth medium (MfGM).
- Immunostaining of satellite cells for Pax7 and MyoD protein
- With a pipette, carefully remove 2 ml of MfGM, leaving remaining 1 ml with myofibers (see Note 13).
- Fix myofibers by adding 1 ml of 4% PFA to each well containing myofibers and incubate for 5 min at room temperature (RT).
- With a pipette, carefully remove MfGM and 4% PFA, leaving myofibers inside the well.
- Add 1 ml of 4% PFA to each well. Incubate the plates for 10 min at RT.
- Carefully remove 4% PFA solution while leaving myofibers inside the well.
- Wash each well containing myofibers with 1 ml of PBS. Incubate for 5 min at RT. Carefully remove PBS leaving myofibers inside the well. Repeat this step 2 times (2 x 5 min).
- Prepare quenching solution.
- Add 2 ml of quenching solution to each well. Incubate for 7 to 10 min at RT.
- Carefully remove quenching solution, leaving myofibers inside the well.
- Wash wells with 1 ml of PBS. Incubate for 5 min at RT. Remove PBS and leave myofibers inside the well. Repeat this step 3 times (3 x 5 min).
- Prepare blocking solution.
- Add 1 ml of blocking solution to each well. Incubate for 60 min at RT.
- Using Pasture pipet, transfer myofibers from the 6-well plate to a 24-well plate (to minimize the quantity of reagents used).
- Make 1:100 dilution of MyoD antibody in blocking solution. Similarly, make 1:5 dilution of Pax7 antibody in blocking solution.
- Carefully remove blocking solution while leaving myofibers inside the well.
- Add 100 µl of each diluted antibody solution per well of the 24-well plate containing myofibers. After this, the final dilution of anti-MyoD will be 1:200 and anti-Pax7 will be 1:10. Incubate the plate overnight at 4 °C.
- Next day, remove primary antibody solution from each well while leaving myofibers inside the well.
- Add 200 μl of PBS to each well of the 24-well plate for washing myofibers. Incubate for 5 min at RT. Remove PBS from the wells and add fresh PBS. Repeat this step 3 times (3 x 5 min).
- Make 1:1,500 dilution of two secondary antibody (i.e., goat anti-rabbit Alexa Fluor 488 conjugate and goat anti-mouse Alexa Fluor 568 conjugate) in blocking solution.
- Add 200 µl of secondary antibodies solution to the well. Incubate for 60 min at RT in the dark.
- Remove secondary antibodies from each well while leaving myofibers inside.
- Add 200 μl of PBS to each well and incubate for 5 min at RT. Carefully remove PBS and leave the myofibers inside the well. Repeat this step 3 times (3 x 5 min).
- Make 1:5,000 dilution of DAPI in PBS.
- Add 200 µl of diluted DAPI solution to each well. Incubate for 3 min at RT in the dark.
- Remove DAPI from each well, leaving myofibers inside.
- Add 200 μl of PBS to each well. Incubate for 5 min at RT. Remove PBS while leaving myofibers inside the well. Repeat this step 2 times (2 x 5 min).
- Finally, add 200 μl of PBS to each well.
- Analyze the myofiber-associated satellite cells for the expression of MyoD (green) and Pax7 (red) under a fluorescent microscope. Representative individual anti-MyoD, anti-Pax7, DAPI stained and merged images generated using this protocol have been published in our recent publications (Hindi and Kumar, 2016; Ogura et al., 2015). A representative image of staining of satellite cell cluster on myofibers after 72 h of culturing is presented in Figure 4.
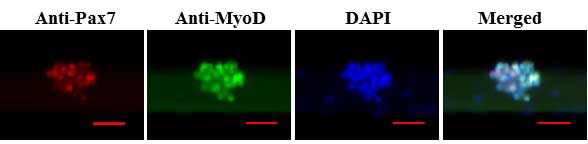
Figure 4. Staining of myofiber-associated satellite cells for Pax7 and MyoD proteins. Representative individual and merged images of myofiber-associated satellite cell cluster stained with anti-Pax7, anti-MyoD, and DAPI following 72 h of culturing. Scale bar = 100 µm.
- With a pipette, carefully remove 2 ml of MfGM, leaving remaining 1 ml with myofibers (see Note 13).
Data analysis
For individual experiments, the sample size should be determined by power analysis. For most of our studies, we have used from 3-5 mice in each group. For analysis of Pax7+ or MyoD+ cells, it is highly recommended that 18-22 myofibers are analyzed from each EDL muscle of each mouse. Data are collected as average number of Pax7+/MyoD-, Pax7+/MyoD+, and Pax7-/MyoD+ cells on each myofibers. These numbers are used to calculate the average from different mice in each group. We present the data as mean ± standard deviation (SD). We used paired or unpaired Student’s t-test to determine statistical differences among different groups similar to as described (Hindi and Kumar, 2016; Ogura et al., 2015; Ogura et al., 2013). A P < 0.05 is considered as statistically significant.
Notes
- It is important to use glass Pasteur pipets in which their ends are smoothened by flame. Sharp end Pasteur pipets can damage the myofibers during picking and handling in subsequent steps.
- Do not add collagenase at this point to preserve enzymatic activity.
- Collagenase aliquots can be prepared ahead of time and stored at -20 °C.
- It is critical to use sterile surgical tools to avoid contamination of cultures especially for long-term analysis.
- Make sure that sharp side of scissors is not facing muscle surface as this could shear and damage the muscle.
- In order to obtain undamaged myofibers, it is necessary to isolate EDL muscle from tendon to tendon during muscle removal process.
- When handling EDL muscle at any step, make sure to hold it by its distal tendons and avoid pinching or grabbing it from the mid-belly.
- Attempting to triturate or release myofibers from EDL before muscle is adequately digested will subject myofibers to mechanical damage because greater trituration force will be required which will reduce the yield of healthy myofibers.
- EDL muscle from wild-type C57BL6 mice get digested within 2 h. However, some other strains may take longer or shorter time for sufficient digestion. It is highly recommended to monitor the digestion of muscle under microscope. Under- or over-digestion of muscle will lead to death and poor yield of myofibers during subsequent steps.
- At this step, do not allow the digested muscle to be aspirated into the Pasture pipet as this could damage the myofibers.
- Once some myofibers have been released into the medium in one well, do not continue the trituration process in the same well to release more myofibers, as previously released myofibers may be damaged during the attempt to dissociate more myofibers from the muscle. For that reason, immediately transfer the muscle to the next well of the tissue culture plate and continue the trituration process to release more myofibers. Continue this process till maximum number of myofibers can be released.
- Healthy myofibers should look translucent with no signs of damage or shearing. Regular striations can be observed on the surface of myofibers under high magnification.
- Removing the entire growth medium before fixing the myofibers may result in bending or shrinking of myofibers, therefore it is advisable to add 4% PFA to myofibers while still in residual amount of growth medium to maintain morphological and structural integrity.
Recipes
- Collagenase II
Prepare 4,000 U/ml stock in ultra-pure water. - Digestion medium
- Prepare base of digestion medium, by supplementing DMEM with 2.5% HEPES and 1% Penicillin (Pen)/streptomycin (Strep) solution. Keep at 37 °C in a water bath till ready for use.
- Once muscle has been isolated and ready for digestion, add collagenase II to make a final concentration of 400 U/ml. Shake briefly by hand.
- Before use, filter digestion medium using a 0.22 µm filter.
Note: For two EDL muscles prepare 5 ml of digestion medium.
- Prepare base of digestion medium, by supplementing DMEM with 2.5% HEPES and 1% Penicillin (Pen)/streptomycin (Strep) solution. Keep at 37 °C in a water bath till ready for use.
- Washing solution
PBS with 1% Pen/Strep - 70% ethanol
Combine 700 ml of 100% ethanol with 300 ml of deionized water. - Basic fibroblast growth factor (bFGF)
Dilute 50 µg of bFGF (50 µg/ml) in 1 ml of 5 mM Tris (pH 7.6) with 0.1% BSA. - Chicken embryo extract (CEE)
Add 10 ml of PBS in a bottle containing lyophilized CEE (50 µg/ml). - Myofiber growth medium (MfGM)
Supplement DMEM with 10% FBS, 1% Pen/Strep, 2% CEE, and 10 ng/ml bFGF.
Warm MfGM at 37 °C in a water bath.
Before use, filter MfGM with a 0.22 µm filter.
Note: For two EDL muscles prepare 36 ml of MfGM. - 4% paraformaldehyde (PFA)
Dilute 0.4 g of PFA in 10 ml of PBS.
Shake vigorously at 60 °C and 250 rpm for 1-2 h until completely dissolved.
Before use, filter 4% PFA with a 0.45 µm filter. - 10% Triton X-100
Combine 100 µl of 100% Triton X-100 with 900 µl of PBS. - Quenching solution
For 10 ml solution, combine 75.07 mg of glycine with 200 µl of 10% Triton X-100 and 50 µl of 5% sodium azide.
Complete to 10 ml with PBS. - Blocking solution
For 10 ml solution, combine 200 mg of BSA with 200 µl of 10% Triton X-100 and 500 µl of horse serum.
Complete to 10 ml with PBS.
Before use, filter blocking solution with a 0.45 µm filter.
Acknowledgments
This work was supported by funding from NIH grants AR059810, AR068313, and AG029623 (to A. Kumar) and AR069985 (to S. M. Hindi).
References
- Anderson, J. E., Wozniak, A. C. and Mizunoya, W. (2012). Single muscle-fiber isolation and culture for cellular, molecular, pharmacological, and evolutionary studies. Methods Mol Biol 798: 85-102.
- Bischoff, R. (1986). Proliferation of muscle satellite cells on intact myofibers in culture. Dev Biol 115(1): 129-139.
- Hindi, S. M., Paul, P. K., Dahiya, S., Mishra, V., Bhatnagar, S., Kuang, S., Choi, Y. and Kumar, A. (2012). Reciprocal interaction between TRAF6 and notch signaling regulates adult myofiber regeneration upon injury. Mol Cell Biol 32(23): 4833-4845.
- Hindi, S. M. and Kumar, A. (2016). TRAF6 regulates satellite stem cell self-renewal and function during regenerative myogenesis. J Clin Invest 126(1): 151-168.
- Kuang, S., Charge, S. B., Seale, P., Huh, M. and Rudnicki, M. A. (2006). Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol 172(1): 103-113.
- Kuang, S. and Rudnicki, M. A. (2008). The emerging biology of satellite cells and their therapeutic potential. Trends Mol Med 14(2): 82-91.
- Keire, P., Shearer, A., Shefer, G. and Yablonka-Reuveni, Z. (2013). Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol Biol 946: 431-468.
- Le Grand, F. and Rudnicki, M. A. (2007). Skeletal muscle satellite cells and adult myogenesis. Curr Opin Cell Biol 19(6): 628-633.
- Ogura, Y., Hindi, S. M., Sato, S., Xiong, G., Akira, S. and Kumar, A. (2015). TAK1 modulates satellite stem cell homeostasis and skeletal muscle repair. Nat Commun 6: 10123.
- Ogura, Y., Mishra, V., Hindi, S. M., Kuang, S. and Kumar, A. (2013). Proinflammatory cytokine tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) suppresses satellite cell self-renewal through inversely modulating Notch and NF-κB signaling pathways. J Biol Chem 288(49): 35159-35169.
- Pasut, A., Jones, A. E. and Rudnicki, M. A. (2013). Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J Vis Exp(73): e50074.
- Rosenblatt, J. D., Lunt, A. I., Parry, D. J. and Partridge, T. A. (1995). Culturing satellite cells from living single muscle fiber explants. In Vitro Cell Dev Biol Anim 31(10): 773-779.
- Shefer, G. and Yablonka-Reuveni, Z. (2005). Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol Biol 290: 281-304.
- Siegel, A. L., Atchison, K., Fisher, K. E., Davis, G. E. and Cornelison, D. D. (2009). 3D timelapse analysis of muscle satellite cell motility. Stem Cells 27(10): 2527-2538.
- Verma, M. and Asakura, A. (2011). Efficient single muscle fiber isolation from alcohol-fixed adult muscle following beta-galactosidase staining for satellite cell detection. J Histochem Cytochem 59(1): 60-67.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gallot, Y. S., Hindi, S. M., Mann, A. K. and Kumar, A. (2016). Isolation, Culture, and Staining of Single Myofibers. Bio-protocol 6(19): e1942. DOI: 10.21769/BioProtoc.1942.
Category
Developmental Biology > Cell growth and fate > Myofiber
Stem Cell > Adult stem cell > Muscle stem cell
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.