- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Measurement of mRNA Decay in Mouse Embryonic Fibroblasts
Published: Vol 6, Iss 13, Jul 5, 2016 DOI: 10.21769/BioProtoc.1858 Views: 12283
Reviewed by: Ivan ZanoniAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2015

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
mRNA stability control is a critical step in the post-transcriptional regulation of gene expression. Actinomycin D, an antibiotic initially used as an anti-cancer drug, has turned out to be a convenient tool for studying the turnover rates of transcripts in cells, due to its inhibition of mRNA synthesis. Here, we describe a protocol for the measurement of mRNA decay after adding actinomycin D into the medium of stable fibroblast cell lines derived from wild-type and tristetraprolin (TTP)-deficient mouse embryonic fibroblast (MEF) cultures, as well as a protocol for determining the relative transcript abundance using semi-quantitative real-time RT-PCR. Northern blotting or NanoString n-Counter are alternative methods to measure mRNA abundance, which is quantified using a phosphorimager in the former case. This protocol is suitable for studying primary cultured cells and stable cell lines derived from transgenic mice and their respective controls, and provides for direct comparisons of mRNA decay rates in otherwise identical cells with and without the gene of interest.
Keywords: MRNA decayMaterials and Reagents
- 60-mm sterile Petri dish (e.g., BD Biosciences, Falcon®, catalog number: 353002 )
Note: Currently, it is “Corning, Falcon®, catalog number: 353002”. - T-75 tissue culture flask (e.g., BD Biosciences, Falcon®, catalog number: 353136 )
Note: Currently, it is “Corning, Falcon®, catalog number: 353136”. - 50 ml sterile conical tube (e.g., BD Biosciences, Falcon®, catalog number: 352070 )
Note: Currently, it is “Corning, Falcon®, catalog number: 352070”. - 384-well microplate (e.g., BioExpress, catalog number: T-6062-1 )
- 1.7 ml RNase-free, DNase-free Posi-Click tubes (Denville Scientific Inc., catalog number: C2170 )
- Mouse wild-type (WT) and TTP-deficient stable fibroblast cell lines (Lai WS et al., 2006)
- 1x Phosphate-buffered saline (PBS) without calcium and magnesium
- 0.05% trypsin/EDTA (Thermo Fisher Scientific, GibcoTM, catalog number: 25300 )
- Fetal bovine serum defined (FBS) (GE Healthcare, HyCloneTM, catalog number: SH30070.03 )
- Dulbecco’s modified Eagle medium (DMEM) (Thermo Fisher Scientific, GibcoTM, catalog number: 11965-092 )
- Penicillin-Streptomycin 10,000 U/ml (Thermo Fisher Scientific, GibcoTM, catalog number: 15140-122 )
- L-glutamine 200 mM (Thermo Fisher Scientific, GibcoTM, catalog number: 25030-081 )
- Recombinant mouse tumor necrosis factor (TNF) (R&D Systems, catalog number: 410-MT )
- Actinomycin D (Sigma-Aldrich, catalog number: A4262 )
- Illustra RNAspin MiniRNA isolation kit (Sigma-Aldrich, GE Healthcare, catalog number: 25-0500-72 )
- SuperScript First-Strand Synthesis System (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18080-051 )
- Power SYBR Green master mix (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4368702 )
- Mercaptoethanol (Sigma-Aldrich, catalog number: M3148 )
- DEPC water (Baltimore Bioworks, catalog number: WA-137-500 )
- Primers for transcripts of interest
- 70% ethanol
- Complete medium (see Recipes)
- Serum-starving medium (see Recipes)
Equipment
- 37 °C, 5% CO2 forced-air incubator (e.g., Thermo Fisher Scientific, FormaTM, model: 3110 )
- Centrifuge with swinging-bucket rotor and adaptors for 50-ml conical tubes
- ABI Prism 7900HT Real-Time PCR System and Sequence Detection System (Applied Biosystems, model: 7900HT ) or similar
- Vortex-Genie-2 (Scientific Industries, catalog number: SI-0236 ) or similar
- Nanodrop 2000c spectrophotometer (Thermo Scientific, model: 2000c )
- Desktop centrifuge (e.g., Eppendorf, model: 5417R )
- DNA Engine Peltier Thermal Cycler (Bio-Rad Laboratories, MJ research, catalog number: PTC-200 ) or similar
- Cell scraper (Corning, Costar, catalog number: 3010 )
Software
- GraphPad Prism software (GraphPad Software, model: version 6.0)
Procedure
- Cell culture
- Mouse stable fibroblast cell lines were derived from MEF cultures from E14.5 TTP KO and littermate WT embryos, as described previously (Lai WS et al., 2006). These stable cell lines have been cultured for more than 200 passages and are well matched in terms of growth rates, morphology, and responses of rapidly inducible genes, such as Fos, to serum stimulation.
- The two stable fibroblast cell lines are maintained in complete medium in T-75 flasks, and passaged every 2-3 days after achieving approximate 70-80% confluence and trypsin treatment.
Note: Cell culture, stimulation and actinomycin D treatment are performed in a Class II Biological Safety Cabinet.
- Mouse stable fibroblast cell lines were derived from MEF cultures from E14.5 TTP KO and littermate WT embryos, as described previously (Lai WS et al., 2006). These stable cell lines have been cultured for more than 200 passages and are well matched in terms of growth rates, morphology, and responses of rapidly inducible genes, such as Fos, to serum stimulation.
- Serum starvation and stimulation
- To plate the cells for experiments, cells are harvested from 3-5 T-75 flasks after washing with 10 ml of PBS followed by trypsinization with 2 ml of trypsin/EDTA and neutralization with 8 ml of complete medium per one T-75 flask, and plated into 60-mm Petri dishes at a density of 2-3 x 105 cells in 5 ml culture medium per Petri dish.
Note: The estimated cell counts harvested from each T-75 flask range between 8 x 105 and 3 x 106 for both the 66 KO and 67 WT stable cell lines, with an average yield of approximate 1.4 x 106 cells. - When the cells reach 70-80% confluence, the cells are washed in serum-free DMEM and then cultured in 5 ml serum-starving medium for at least 16 h of serum starvation.
- Add recombinant murine TNF (or other stimuli) into serum-starving medium for a final TNF concentration of 10 ng/ml, and then harvest cells at various time points after treatment.
- To plate the cells for experiments, cells are harvested from 3-5 T-75 flasks after washing with 10 ml of PBS followed by trypsinization with 2 ml of trypsin/EDTA and neutralization with 8 ml of complete medium per one T-75 flask, and plated into 60-mm Petri dishes at a density of 2-3 x 105 cells in 5 ml culture medium per Petri dish.
- Actinomycin D treatment
- Actinomycin D solution is prepared by dissolving the powder in DEPC water. The solution is stored at 4 °C in the dark for a final concentration of 2-5 mg/ml.
Note: It often takes at least 1-2 days for the actinomycin D to get fully dissolved in water at 4 °C; gently invert the bottle several times to mix the solution prior to use. DMSO is not used as a solvent in this protocol. - After 30 min or other times of TNF treatment of fibroblast cells (10 ng/ml in this example), or treatment with other stimuli, actinomycin D is added directly to the serum-starving medium for a final concentration of 5-10 μg/ml; TNF and other stimuli are not removed at the time of actinomycin D addition.
Note: We routinely perform a time course study first for an individual stimulus to determine the kinetic expression pattern of the gene of interest. The time point at which cells express the highest levels of the transcript of interest is then usually chosen as the time for actinomycin D addition. - Cells are then harvested at 0, 10, 20, 30, 45, 60, 90, and 120 min after the addition of actinomycin D, or other times as indicated; each sample is comprised of pooled cells from three 60-mm dishes.
Note: It is not recommended to use trypsin/EDTA for cell harvest followed by RNA extraction. It is necessary to proceed immediately to Procedure D by adding sample lysis buffer directly into the Petri dishes after one rinse with ice-cold PBS.
- Actinomycin D solution is prepared by dissolving the powder in DEPC water. The solution is stored at 4 °C in the dark for a final concentration of 2-5 mg/ml.
- RNA extraction, reverse transcription and semi-quantitative real-time PCR
- To prepare cells for RNA extraction, the culture medium is aspirated and the cells are washed once in 5 ml of ice-cold PBS per one 60-mm dish. RA1 lysis buffer (from the Illustra RNAspin MiniRNA isolation kit) supplemented with freshly added mercaptoethanol (ME, 1:100 dilution) is added directly into the Petri dishes, and the lysates are then scraped off using cell scrapers. The lysates are then pooled from triplicate plates and transferred into a new fresh 1.7 ml microcentrifuge tube. This procedure can be performed on the bench at room temperature.
Note: For cells in suspension other than adherent cells such as fibroblasts, samples are harvested first by centrifugation, and then washed once in PBS before adding RA1 lysis buffer with ME. - Follow the manufacturer’s instructions in the GE Healthcare illustra RNAspin MiniRNA isolation kit for total RNA extraction; this includes steps of lysate vortexing, filtering through the shredder column to decrease the viscosity of lysates, and on-column digestion with RNase-free DNase I. Lysates that have flowed through the shredder column can be transferred into a new tube and stored at -80 °C.
- Take 1 μl of RNA from each sample to check for RNA quantity and quality with Nanodrop.
Note: The expected RNA yield from three combined dishes in this protocol is around 16 µg. - 750 ng RNA is used to synthesize first-strand cDNAs using oligo (dT)12-18 primers and SuperScript III Reverse Transcriptase (Invitrogen) as per manufacturer’s protocol.
- cDNA is then diluted to 1.5-2 ng/µl with DEPC water, and is ready for use or can be stored at -20 °C.
- Real-time PCR is performed using SYBR Green master mix and the ABI Prism 7900 Sequence Detection System in a 384-well plate. Each reaction is comprised of 1x SYBR Green master mix, 1-2 ng cDNA, 250 nM of each primer in a total of 10 μl reaction volume, i.e., 5 µl of 2x SYBR Green master mix, 0.5 µl of each forward and reverse primer at 5 µM, 1 µl of cDNA, and 3 µl of DEPC water per reaction. Each plate contains “no template” controls for individual transcripts as well as housekeeping transcripts such as Actb mRNA for every sample as an internal control.
- Results are analyzed using the ΔΔCt method (Pfaffl MW 2001). Ct values from duplicate or triplicate samples are first normalized to their respective internal housekeeping transcripts, Actb mRNA in this example, and then normalized to their respective samples before the addition of actinomycin D, which are set at 1. The results are expressed as percentages of mRNA abundance relative to time 0. A representative experiment is shown in Table 1.
Note: It is critical to validate primer amplification efficiency and specificity prior to use. To check primer amplification efficiency, a 10-fold serial dilution of cDNA across 4-5 log range is used. The relative Ct values after normalization to an internal housekeeping transcript are plotted against the concentrations at log values for linear regression curves. Any absolute values of slopes less than or equal to 0.1 are considered to pass the efficiency test when paired with the housekeeping gene in the assay. A representative experiment is shown in Figure 1.
Table 1. Calculation of the Relative Transcript Abundance using the ΔΔCt Method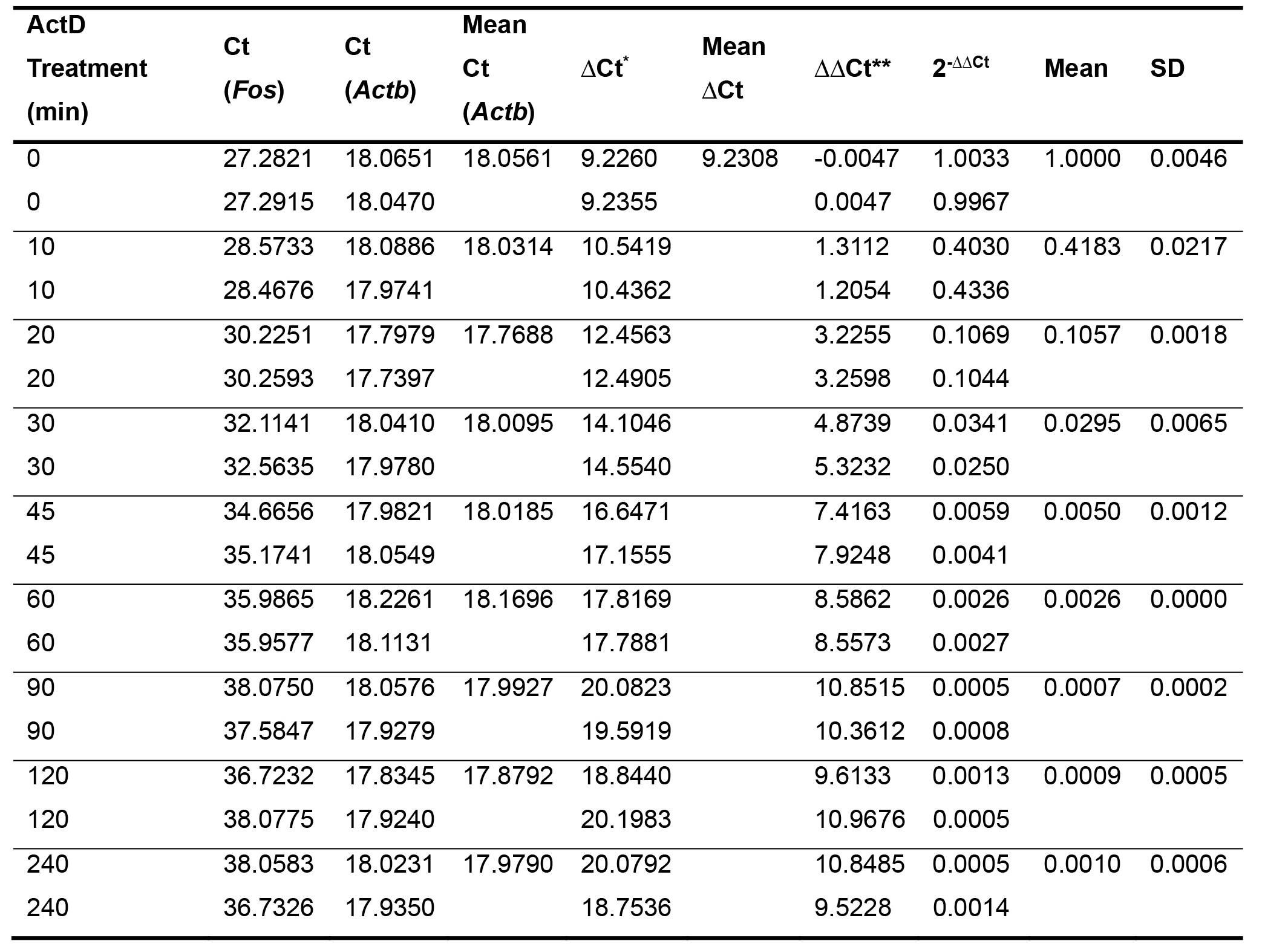
*∆Ct = Ct (Fos, column 2) – Ave Ct (Actb, column 4); **∆∆Ct = ∆Ct (column 5)– Ave ∆Ct at time 0 (column 6). Actinomycin D (ActD) was added after 30 min of TNF stimulation. - The transcript turnover rates are then calculated based on the non-linear fit one phase exponential decay curves using GraphPad Prism software, and are generally expressed as times to 50% mRNA decay for each experiment. A representative experiment is shown in Figure 2.
- To prepare cells for RNA extraction, the culture medium is aspirated and the cells are washed once in 5 ml of ice-cold PBS per one 60-mm dish. RA1 lysis buffer (from the Illustra RNAspin MiniRNA isolation kit) supplemented with freshly added mercaptoethanol (ME, 1:100 dilution) is added directly into the Petri dishes, and the lysates are then scraped off using cell scrapers. The lysates are then pooled from triplicate plates and transferred into a new fresh 1.7 ml microcentrifuge tube. This procedure can be performed on the bench at room temperature.
Representative data
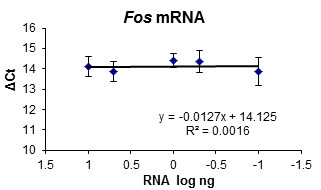
Figure 1. Validation of real-time PCR primers. Serial dilutions of cDNA at 10, 5, 1, 0.5, 0.1, and 0.01 ng were prepared as templates for real-time PCR analysis using primers against Fos and Actb transcripts. The X-axis in the graph shown is the log value of cDNA; ∆Ct shown on the Y-axis are the differences in the Ct values between Fos and Actb mRNAs. The data presented are means ± SD of duplicates from one representative experiment. A linear regression curve was plotted with an absolute slope value of ≤0.1. In this example, the Ct value for the Fos transcript at 0.01 ng was undetectable, so it was not included in the calculation of the regression curve.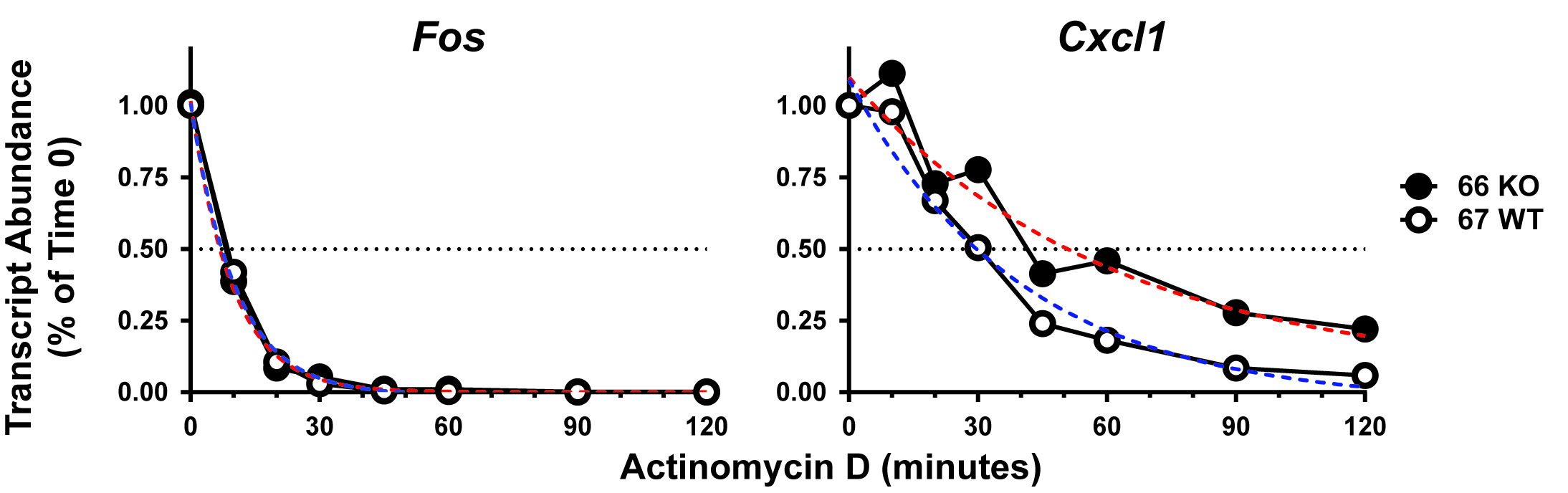
Figure 2. Stability of Fos and Cxcl1 transcripts after TNF stimulation in the presence and absence of TTP. Actinomycin D, to inhibit transcription, was added after 30 min of TNF treatment of serum-starved fibroblast cell lines. The cells were harvested for total RNA extraction at the indicated times, and mRNA levels were measured with real-time RT-PCR. Transcript concentrations were normalized to those of the Actb transcripts, and were expressed as fractions of abundance in the TNF-treated samples prior to the addition of actinomycin D. The results shown are means of replicate samples from one representative experiment. The transcript turnover rates were calculated based on the non-linear fit one phase exponential decay curves using GraphPad software (red dotted lines, 66 KO cells; blue dotted lines, 67 WT cells), and expressed as times to 50% mRNA decay for each experiment (the time points when the dotted black lines for the 50% mRNA decay lines cross the red and blue dotted lines, respectively, for each curve).
Recipes
- Complete medium
DMEM
10% FBS
1% Pen-Strep
2 mM glutamine - Serum-starving medium
DMEM
0.5% FBS
1% Pen-Strep
2 mM glutamine
Acknowledgments
This protocol was adapted from previously published studies, Lai et al. (2006) and Horner et al. (2009), and was used as described here in Qu et al. (2015). We thank Drs. Melissa Wells and Diana Cruz-Topete for comments on the protocol. This research was supported by the Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health.
References
- Horner, T. J., Lai, W. S., Stumpo, D. J. and Blackshear, P. J. (2009). Stimulation of polo-like kinase 3 mRNA decay by tristetraprolin. Mol Cell Biol 29(8): 1999-2010.
- Lai, W. S., Parker, J. S., Grissom, S. F., Stumpo, D. J. and Blackshear, P. J. (2006). Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol Cell Biol 26(24): 9196-9208.
- Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9): e45.
- Qiu, L. Q., Lai, W. S., Bradbury, A., Zeldin, D. C. and Blackshear, P. J. (2015). Tristetraprolin (TTP) coordinately regulates primary and secondary cellular responses to proinflammatory stimuli. J Leukoc Biol 97(4): 723-736.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Qiu, L. Q., Lai, W. S., Stumpo, D. J. and Blackshear, P. J. (2016). Measurement of mRNA Decay in Mouse Embryonic Fibroblasts. Bio-protocol 6(13): e1858. DOI: 10.21769/BioProtoc.1858.
Category
Immunology > Immune cell function > General
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.