- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Small-scale Subcellular Fractionation with Sucrose Step Gradient

Published: Vol 4, Iss 11, Jun 5, 2014 DOI: 10.21769/BioProtoc.1138 Views: 17523
Reviewed by: Fanglian He
Original research article
The authors used this protocol in:

Jun 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Here, we introduce the protocol for small-scale and simple subcellular fractionation used in our recent publication (Taguchi et al., 2013), which uses homogenization by passing through needles and sucrose step-gradient.
Subcellular fractionation is a very useful technique but usually a large number of cells are required. Because we needed subcellular fractionation of transiently-transfected cells, we developed a protocol for smaller numbers of cells. Our protocol for the subcellular fractionation is based on the protocol published by de Araújo and Huber (de Araujo et al., 2007), although substantial modifications have been made according to our experiences and information from personal communications. As optimal conditions seem to vary between cell lines, we advise to further modify the protocol to optimize for individual experiments. Our method is simple but sufficient for analysis of integral membrane proteins or proteins anchored to organelles by glycosylphosphatidylinositol or other lipid anchors, e.g. prion protein. However, proteins non-covalently attached to membranes or membrane proteins of organelles seem to be more prone to dissociation from the organelles during preparation and, if these proteins are the object of study, further modifications might be necessary.
Unlike in a continuous gradient, where a protein of interest is scattered over a wide range, step-gradient fractionation is advantageous in detection of relatively small amounts of proteins from small-scale experiments, because it concentrates the protein of interest in one fraction, if an appropriate combination of sucrose concentrations is used.
Materials and Reagents
- Neuro2a cells (N2a)
- Sucrose (Sigma-Aldrich, catalog number: S9378-500G )
- 1 M Tris-HCl (pH 7.1)
- 0.5 M EDTA (Millipore, catalog number: 324503-1KG )
- Purified water
- 100 mg/ml solution of cycloheximide in DMSO (Sigma-Aldrich, catalog number: C4859-1ML )
- Complete protease-inhibitor cocktail (Roche Diagnostics, catalog number: 04693116001 )
- OptiMEM I supplemented with 10% fetal bovine serum
- Phosphate-buffered saline without calcium/magnesium (Ca/Mg) (Life Technologies, catalog number: 10010-023 )
- Deoxycholic acid (Sigma-Aldrich, catalog number: D2510-100G )
- Triton X-100 (Sigma-Aldrich, catalog number: 93443-100ML )
- Sodiumdodecyl sulfate (Sigma-Aldrich, catalog number: L6026-50G )
- Glycerol (Sigma-Aldrich, catalog number: G9012-500ML )
- Phosphate-buffered 0.5% Triton X-100 (TX100)/0.5% deoxycholate (DOC) lysis buffer (see Recipes)
- 5x sample buffer (see Recipes)
Equipment
- 6-well plate
- 1 ml BD Luer-LokTM disposable syringe (BD, catalog number: 309628 )
- 25G ultra-thin-wall needle (Terumo Medical Corporation, catalog number: NN-2525R )
- Cell scraper
- Centrifuge tube (thinwall, Ultra-ClearTM, 5 ml, 13 x 51 mm) (Beckman Coulter, catalog number: 344057 )
- Ultracentrifuge (Beckman Coulter, model: L8-80M )
- Pre-chilled swing bucket rotor (Beckman Coulter, model: SW50.1 )
- Portable refractometer (optional) (e.g. ATAGO, model: PAL-1 )
- Inverted microscope
Procedure
- Preparation of sucrose and homogenization buffers
- Decide the sucrose concentrations of the layers for the step gradient, e.g. 36%, 33% and 8.5% (Taguchi et al., 2013).
- The concentration of sucrose buffers is based on “percent by weight”, i.e. sucrose concentration (%) = 100 x sucrose (g)/[sucrose (g) + water (g)].
- Then, take necessary amounts of sucrose and dissolve it in purified water. Usually, measuring the actual concentration is not necessary as long as the amounts of sucrose and water are correct. If necessary, we confirm the actual concentration with a portable refractometer.
- After preparation of the sucrose solution in water, we measure the volume and add 1/250-volume of 1 M Tris-HCl (pH 7.1) to make the final concentration of 4 mM. We use Tris-HCl buffer but the buffer type should be determined based on the following procedures and the purpose of the experiments.
- For the homogenization buffer, the sucrose concentration of the homogenization buffer is 8.5% and, in addition to the 4 mM Tris-HCl, we add EDTA to a final concentration 2 mM, and cycloheximide to 30 µg/ml, and complete protease inhibitor cocktail with a dosage according to the manufacturer’s instruction. Cycloheximide is added to maintain the polysomes on the endoplasmic reticulum and to increase the density of the organelles for more efficient separation (de Araujo et al., 2007).
- Decide the sucrose concentrations of the layers for the step gradient, e.g. 36%, 33% and 8.5% (Taguchi et al., 2013).
- Homogenization of cells
- Rinse the N2a cells, 100% confluent in OptiMEM I supplemented with 10% fetal bovine serum on a well of a 6-well plate, once with pre-chilled PBS without Ca/Mg, then add 700 µl of 3 mM EDTA in PBS and incubate for a few minutes until cells can be easily detached from the bottom by pipetting.
- Although N2a cells can be detached with this procedure, more adhesive cells might require mechanical detachement with cell scrapers. On the other hand, less-adhessive cells might not need EDTA in PBS for detachment.
- Thoroughly detach the cells and transfer the cells in PBS into pre-chilled 1.5 ml tubes. Then, rinse the bottom of the well with 500 µl of 3 mM EDTA in PBS and add it to the tube.
- Keep cells and tubes always on ice.
- Although N2a cells can be detached with this procedure, more adhesive cells might require mechanical detachement with cell scrapers. On the other hand, less-adhessive cells might not need EDTA in PBS for detachment.
- Centrifuge the cell suspension at 200 x g at 4 °C for 5 min and then carefully remove the supernatant. Resuspend the pelleted cells in 4-7 volumes of homogenization buffer. For cells from one well of a 6 well plate, we usually add 200 µl of homogenization buffer.
- (Optional) Before homogenization, take 10 µl of cell suspension for whole-cell lysate analysis and mix with 30 μl of phosphate-buffered 0.5% Triton X-100/0.5% deoxycholate lysis buffer.
- After incubation on ice for 10 min, centrifuge to precipitate the cell debris and carefully aspirate the supernatant as whole-cell lysate.
- Add 10 μl of 5x sample buffer and boil for 10 min to prepare "whole-cell lysate" samples without fractionation.
- After incubation on ice for 10 min, centrifuge to precipitate the cell debris and carefully aspirate the supernatant as whole-cell lysate.
- Homogenize the rest of the cells by passing through a 25G ultra-thin-wall needle attached on a 1 ml-syringe until ~90% of cells are disrupted. As extent of destruction of cells is critical for yields of organelles and proteins attached to them, this step has to be carefully evaluated.
- At first, checking the status of cells after every 10-15 strokes is recommended. This is done by taking 1-2 µl of homogenate and suspending it into a drop of PBS on the lid of a 6-well plate and observing it on an inverted microscope. Intact naked nuclei which are released from the broken cells are observed as relatively flat and small rounded structures, whereas unbroken cells are obviously larger and more spherical.
- How many strokes are necessary for sufficient disintegration of cells can vary between cell types. For example, the cells we are using require more than 30 strokes.
- As a note of caution, producing air bubbles should be avoided during the homogenization procedure.
- At first, checking the status of cells after every 10-15 strokes is recommended. This is done by taking 1-2 µl of homogenate and suspending it into a drop of PBS on the lid of a 6-well plate and observing it on an inverted microscope. Intact naked nuclei which are released from the broken cells are observed as relatively flat and small rounded structures, whereas unbroken cells are obviously larger and more spherical.
- After cells are sufficiently disintegrated, centrifuge the homogenate at 2,000 x g for 5 min at 4 °C. Carefully take the supernatant as the "post-nuclear fraction" and estimate the volume by marking the level of the homogenate in the pipet tip and measuring the volume of water which fills up to the mark.
- Add 1.4 volume of 62% sucrose buffer and mix well until the mixture becomes completely homogeneous. As the two solutions do not easily mix, patienly mix by gentle pipetting with a ‘genomic tip’ (or a pipet tip with tip cut-off) and stirring with the tip at the same time to a point where pipetting does not make "clear streaks" in the solution (de Araujo et al., 2007).
- Keep the post-nuclear fraction chilled throughout the procedure.
- Add 1.4 volume of 62% sucrose buffer and mix well until the mixture becomes completely homogeneous. As the two solutions do not easily mix, patienly mix by gentle pipetting with a ‘genomic tip’ (or a pipet tip with tip cut-off) and stirring with the tip at the same time to a point where pipetting does not make "clear streaks" in the solution (de Araujo et al., 2007).
- Place the post-nuclear fraction mixed with 62% sucrose buffer at the bottom of the centrifuge tube and overlay with three layers of sucrose solutions, e.g. 36%, 33% and finally homogenization buffer (8.5%), 1.5 ml each, from bottom to top (Taguchi et al., 2013).
- Carefully pour the sucrose buffer not to agitate the interphase too much. Tilt the tube, put the point of the pipette tip just above the surface of the lower layer in the tube and carefully push out the solution in the tip to let it slowly float along the wall.
- Intervals between interfaces should be distant enough to avoid contamination from adjacent interfaces during the procedure of harvesting the concentrated organelles.
- As optimal sucrose concentrations of the layers for separation of the organelles of interest seem to vary between cell lines, modify the combination of sucrose concentrations according to the distribution of organelle marker proteins until the most suitable combination is found.
- Carefully pour the sucrose buffer not to agitate the interphase too much. Tilt the tube, put the point of the pipette tip just above the surface of the lower layer in the tube and carefully push out the solution in the tip to let it slowly float along the wall.
- Ultra-centrifuge the gradient at 40,000 rpm at 4 °C for 1.5 h. We used a Beckman SW50.1 swing rotor and a Beckman L8-80M ultracentrifuge.
- Upon ultra-centrifugation, fractionated organelles are condensed between the layers and visible as “milky-white” bands in interfaces (Figure 1). As the sample presented in the figure was prepared from a 100 mm-dish, the “milky-white” bands are more clearly visible. With N2a cells confluent on a well of 6-well plate, the bands are visible, but less pronounced. Aspirate the "milky bands" as thoroughly as possible. This requires scanning the pipet tip, gradually aspirating, right over a white band in the interface, until it is completely collected. We usually take more volume so that we can most efficiently recover the desired organelles, e.g. 350 µl.
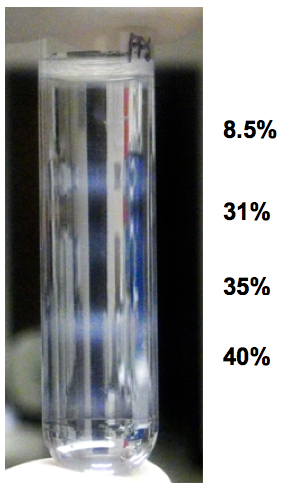
Figure 1. Example of appearance of fractionated organelles in step-gradient fractionation. The sample was from a 100 mm-cell culture dish and four layers were overlaid on the homogenate. Note that there are four "milky-white" bands at the interfaces of the five layers.
- If the purpose is just to evaluate distribution of a specific protein of interest in the fractions, we usually dilute the collected interfaces with 300 μl of PBS with 30 μg of bovine-serum albumin as a carrier protein, and then subject this to methanol/chloroform precipitation (Taguchi et al., 2013). Dissolve the proteins in 1x sample buffer and boil for 10 min to make "fractionated" samples.
- Rinse the N2a cells, 100% confluent in OptiMEM I supplemented with 10% fetal bovine serum on a well of a 6-well plate, once with pre-chilled PBS without Ca/Mg, then add 700 µl of 3 mM EDTA in PBS and incubate for a few minutes until cells can be easily detached from the bottom by pipetting.
Recipes
- Phosphate-buffered 0.5% Triton X-100 (TX100)/0.5% deoxycholate (DOC) lysis buffer
- First prepare 5% TX100/DOC stock solution
Triton X-100
5 ml
Deoxycholic
5 g
Purified water
up to 100 ml
- Then mix 5% TX100/5% DOC, 10x phosphate-buffered saline (PBS) and water in a 50 ml-conical tube
5% TX100/5% DOC
5 ml
10x PBS
5 ml
Purified water
up to 50 ml
- First prepare 5% TX100/DOC stock solution
- 5x sample buffer
SDS
1.2 g
1 M Tris-HCl (pH7.1)
2.5 ml
Glycerol
4 ml
0.5% BPB
300-500 μl
Water
up to 10 ml
Acknowledgments
This protocol was adapted from Taguchi et al. (2013). This work was supported by grants for the National Institute of Health R01 NS076853-01A1 and the Alberta Prion Research Institute (AB, Canada).
References
- de Araujo, M. E. and Huber, L. A. (2007). Subcellular fractionation. Methods Mol Biol 357: 73-85.
- Taguchi, Y., Mistica, A. M., Kitamoto, T. and Schätzl, H. M. (2013). Critical significance of the region between Helix 1 and 2 for efficient dominant-negative inhibition by conversion-incompetent prion protein. PLoS Pathog 9(6): e1003466.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Taguchi, Y. and Schätzl, H. M. (2014). Small-scale Subcellular Fractionation with Sucrose Step Gradient. Bio-protocol 4(11): e1138. DOI: 10.21769/BioProtoc.1138.
- Taguchi, Y., Mistica, A. M., Kitamoto, T. and Schätzl, H. M. (2013). Critical significance of the region between Helix 1 and 2 for efficient dominant-negative inhibition by conversion-incompetent prion protein. PLoS Pathog 9(6): e1003466.
Category
Cell Biology > Organelle isolation > Fractionation
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.