- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Low-input Capture-C: A Chromosome Conformation Capture Assay to Analyze Chromatin Architecture in Small Numbers of Cells
Published: Vol 7, Iss 23, Dec 5, 2017 DOI: 10.21769/BioProtoc.2645 Views: 12820
Reviewed by: Jyotiska ChaudhuriAlexandros AlexandratosAnonymous reviewer(s)
Original research article
The authors used this protocol in:

2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Chromosome conformation capture (3C) techniques are crucial to understanding tissue-specific regulation of gene expression, but current methods generally require large numbers of cells. This protocol describes two new low-input Capture-C approaches that can generate high-quality 3C interaction profiles from 10,000-20,000 cells, depending on the resolution used for analysis.
Keywords: Nuclear organizationBackground
3C techniques play a key role in investigating how nuclear organization and structural interactions between regulatory elements relate to gene activity (Dekker et al., 2002). As these interactions are highly tissue-specific, it is crucial that 3C experiments are performed in well defined, purified cell populations.
A major limitation of 3C techniques is the large numbers of cells required: current methods use between 100,000 and 10 million cells (Davies et al., 2017). Many primary tissues and rare cell populations are not available in these numbers. We have therefore developed two new low-input Capture-C approaches that can generate high-quality interaction profiles from ~20,000 cells at maximum resolution (individual DpnII fragments), and from ~10,000 cells using windowing based analysis (Oudelaar et al., 2017).
Materials and Reagents
- 3C library preparation
- LowBind DNA 1.5 ml microtube
- Phase Lock Gel (PLG) Light tubes (VWR, Quantabio, catalog number: 733-2477 )
- Genomic DNA ScreenTape (Agilent Technologies, catalog number: 5067-5366 )
- Tapestation Loading Tips (Agilent Technologies, catalog number: 5067-5153 )
- 1 M glycine (Sigma-Aldrich, catalog number: G7126 )
- Phosphate-buffered saline (PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10010031 )
- Growth media
- 37% formaldehyde (Sigma-Aldrich, catalog number: 252549 )
- Ethanol absolute > 99.8% (VWR, catalog number: 20821.330 , or equivalent)
- Dry ice or liquid nitrogen
- PCR grade water (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9932 )
- SDS (20% v/v in water) (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9820 )
- Triton X-100 (20% v/v in water) (Sigma-Aldrich, catalog number: T8787 )
- DpnII (50,000 U/ml) (New England Biolabs, catalog number: R0543M )
- T4 DNA ligase (30 U/μl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EL0013 )
- Proteinase K (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EO0491 )
- RNase (DNase-free) (Roche Diagnostics, catalog number: 11119915001 )
- Phenol-chloroform-isoamyl alcohol (PCI) 25:24:1 (Sigma-Aldrich, catalog number: 77617 )
- 3 M NaOAc (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9740 )
- GlycoBlue (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9515 )
- Genomic DNA Reagents (Agilent Technologies, catalog number: 5067-5365 )
- TaqMan Universal PCR Master Mix II without UNG (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4440040 )
- KAPA Sybr Fast Universal (Kapa Biosystems, catalog number: KK4602 )
- Fresh lysis buffer (see Recipes)
- Tris pH 8 (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9855G )
- Sodium chloride (NaCl) (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9760G )
- Igepal CA-630 (100 μl of 10%) (Sigma-Aldrich, catalog number: I8896 )
- cOmplete Protease Inhibitor Cocktail (1 tablet in 2 ml of PCR grade water for 25x; store at -20 °C) (Sigma-Aldrich, Roche Diagnostics, catalog number: 11873580001 )
- PCR grade water
- Tris pH 8 (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9855G )
- qPCR primers (Appendix I)
- LowBind DNA 1.5 ml microtube
- Addition of Illumina sequencing adaptors by ligation or tagmentation to generate Capture-C libraries
LI-Capture-C- Covaris microTUBE AFA Fiber pre-split snap-cap 6 x 16 mm (Covaris, catalog number: 520045 )
- LowBind DNA 1.5 ml microtube
- PCR tube
- D1000 ScreenTape (Agilent Technologies, catalog number: 5067-5582 )
- TapeStation Loading Tips (Agilent Technologies, catalog number: 5067-5153 )
- NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, catalog number: E7370S/L )
- Agencourt Ampure XP SPRI Beads (Beckman Coulter, catalog number: A63881 )
- NEBNext Multiplex Oligos for Illumina Primer set 1 (New England Biolabs, catalog number: E7335S/L )
- NEBNext Multiplex Oligos for Illumina Primer set 2 (New England Biolabs, catalog number: E7500S/L )
- Herculase II Fusion Polymerase Kit (Agilent Technologies, catalog number: 600677 )
- PCR grade water (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9932 )
- Ethanol absolute > 99.8% (VWR, catalog number: 20821.330 , or equivalent)
- D1000 Reagents (Agilent Technologies, catalog number: 5067-5583 )
- Qubit dsDNA BR Assay kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32850 )
Tag-Capture-C- LowBind DNA 1.5 ml microtube
- PCR tube
- High Sensitivity D1000 ScreenTape (Agilent Technologies, catalog number: 5067-5584 )
- D1000 ScreenTape (Agilent Technologies, catalog number: 5067-5582 )
- TapeStation Loading Tips (Agilent Technologies, catalog number: 5067-5153 )
- Nextera DNA Sample Preparation Kit (Illumina, catalog number: FC-121-1030 / 1031 )
- Zymo DNA Clean & Concentrator-5 Kit (Zymo Research, catalog number: D4013 )
- KAPA HiFi PCR Kit with dNTP (KK2102) (Roche Diagnostics, catalog number: 07958846001 )
- Custom designed index primers
- Agencourt Ampure XP SPRI Beads (Beckman Coulter, catalog number: A63881 )
- PCR grade water (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9932 )
- Ethanol absolute > 99.8% (VWR, catalog number: 20821.330 , or equivalent)
- High Sensitivity D1000 Reagents (Agilent Technologies, catalog number: 5067-5585 )
- D1000 Reagents (Agilent Technologies, catalog number: 5067-5583 )
- Qubit dsDNA BR Assay kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32850 )
- Covaris microTUBE AFA Fiber pre-split snap-cap 6 x 16 mm (Covaris, catalog number: 520045 )
- Enrichment for viewpoints of interest by oligonucleotide capture
- Safeseal Microcentrifuge Tubes (Sorenson BioScience, catalog number: 39640T )
- D1000 ScreenTape (Agilent Technologies, catalog number: 5067-5582 )
- Tapestation Loading Tips (Agilent Technologies, catalog number: 5067-5153 )
- Nimblegen SeqCap EZ Hybridization and wash kit (Roche Diagnostics, catalog number: 05634261001 )
- Nimblegen SeqCap EZ Accessory kit v2 (Roche Diagnostics, catalog number: 07145594001 )
- 1 μg/μl COT DNA of relevant species (Mouse) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18440016 )
- M-270 Streptavidin Dynabeads (Thermo Fisher Scientific, InvitrogenTM, catalog number: 65305 )
- Agencourt Ampure XP SPRI Beads (Beckman Coulter, catalog number: A63881 )
- PCR grade water (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9932 )
- Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32850 )
- Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32851 )
- D1000 Reagents (Agilent Technologies, catalog number: 5067-5583 )
- KAPA Library Quantification Complete Kit (Universal) (Kapa Biosystems, catalog number: KK4824 )
- LI-Capture-C
Nimblegen SeqCap EZ HE-oligo kit A (Roche Diagnostics, catalog number: 06777287001 )
Nimblegen SeqCap EZ HE-oligo kit B (Roche Diagnostics, catalog number: 06777317001 ) - Tag-Capture-C
xGen Custom Blocking Oligos (P5/i5 & P7/i7 Nextera blocker)
- Safeseal Microcentrifuge Tubes (Sorenson BioScience, catalog number: 39640T )
Equipment
- Centrifuge
- Incubator (any)
- Eppendorf Thermomixer C (or equivalent shaking incubator) (Eppendorf, model: ThermoMixer® C , catalog number: 5382000015)
- P10, P20, P200 and P1000 pipettes (any)
- Thermocycler (any)
- NanoDrop
- Agilent 2200 TapeStation (Agilent Technologies, model: Agilent 2200 TapeStation System )
Note: Assessments on the Agilent TapeStation can also be performed on the Agilent BioAnalyzer. - Sonicator (Covaris, model: S220 Focused-ultrasonicator)
- DynaMag-2 (Thermo Fisher Scientific, catalog number: 12321D )
- Vacuum Concentrator (SciQuip, CHRIST series)
Procedure
Overview
Note: Overview of the procedure (Graphics adapted from Davies et al., 2017).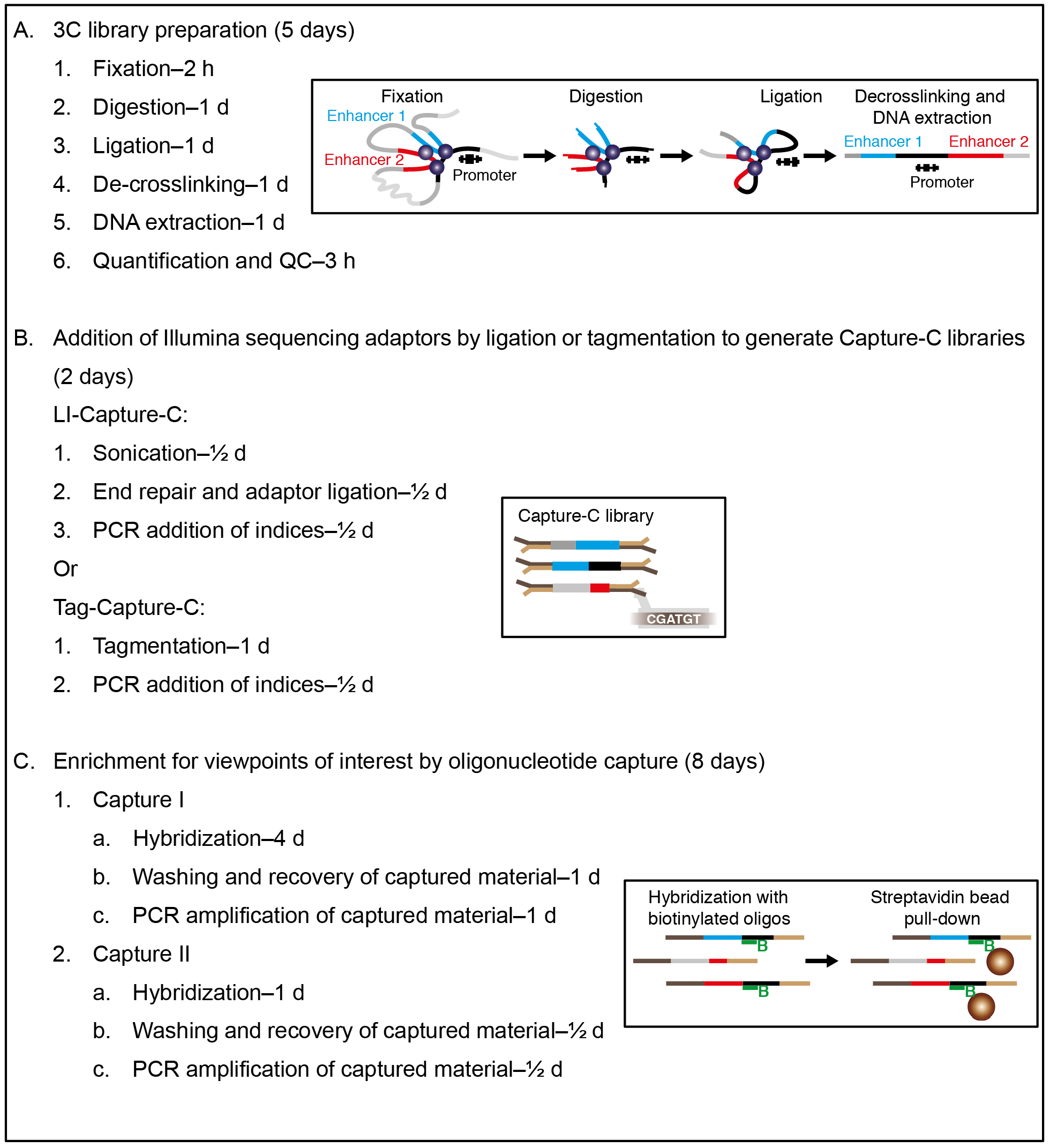
Steps
- 3C library preparation
- Fixation (2 h)
Note: The fixation procedure described below has been optimized for erythroid cells. Depending on the cell type used, some of centrifugation steps might need to be adjusted to ensure proper pelleting.- Pre-cool centrifuge to 4 °C. Chill glycine, PBS, and lysis buffer (see Recipes).
- Collect cells from tissue and make single-cell suspensions of 10,000 to 106 cells in 1 ml growth media. If sorting cells, sort into 100 μl media and add media afterwards to a total volume of 1 ml.
- Add 54 μl 37% formaldehyde (2% final concentration), mix well and incubate for 10 min at RT on a rocking or tumbling incubator.
- Quench by adding 150 μl 1 M cold glycine (1/8 of the final volume. i.e., 1.2 ml).
- Centrifuge for 15 min/500 x g/4 °C.
- Carefully remove supernatant, do not disturb pellet. Can leave ~5% behind.
- Wash pellet by gently resuspending in 1 ml cold PBS.
- Centrifuge for 15 min/500 x g/4 °C.
- Carefully remove supernatant, do not disturb pellet. Can leave ~5% behind.
- Resuspend pellet in 500 μl cold lysis buffer.
- Incubate for 20 min on ice.
- Snap freeze with ethanol and dry ice or liquid nitrogen. Snap freezing aids digestion so cells can be thawed again for digestion at this point or stored long-term at -80 °C.
SAFE STOPPING POINT: Store at -80 °C.
- Pre-cool centrifuge to 4 °C. Chill glycine, PBS, and lysis buffer (see Recipes).
- Digestion (1 day)
- Pre-warm a ThermoMixer to 37 °C.
- Prepare Digestion Mix (make a master mix for multiple libraries):
PCR grade water 152.5 μl 10x DpnII Buffer 20 μl 20% SDS 2.5 μl - Defrost aliquots of 10,000 to 106 formaldehyde-fixed cells for each reaction.
- Centrifuge for 10 min/15,000 x g/4 °C.
- Carefully remove all the lysis buffer, using a P20 to remove residual buffer without disturbing the pellet.
- Resuspend in 175 μl of Digestion Mix.
- Shake horizontally (1,400 rpm) for 1 h at 37 °C using the ThermoMixer.
- Add 16 μl of 20% Triton X-100 (1.7% final concentration) and shake for 1 h at 37 °C.
- Add first aliquot of 3 μl of DpnII enzyme and shake at 37 °C for at least two hours.
- Add a second aliquot of 3 μl of DpnII enzyme at the end of the day and shake overnight.
- Add a further 3 μl of DpnII enzyme the next morning and shake for at least three more hours.
- Pre-warm a ThermoMixer to 37 °C.
- Ligation (1 day)
- Place the digests on the 65 °C block for 20 min to heat inactivate the restriction enzyme.
- Cool the digest on ice and cool the ThermoMixer to 16 °C.
- Make Ligation Mix (make a master mix for multiple libraries):
PCR grade water 125 μl 10x ligation buffer 36 μl - Add 161 μl Ligation Mix to digest.
- Add 4 μl T4 ligase and shake at 300 rpm/16 °C for ~22 h.
- Place the digests on the 65 °C block for 20 min to heat inactivate the restriction enzyme.
- De-crosslinking (1 day)
Add 2 μl Proteinase K (20 mg/ml) and incubate at 65 °C overnight. - DNA extraction (1 day)
- Add 4 μl of RNase to the ligation reaction and incubate at 37 °C for 30 min.
- Add 350 μl phenol-chloroform-isoamyl alcohol and vortex thoroughly.
- Transfer to a 2 ml PLG Light tube and spin for 5 min/15,000 x g/RT.
- Transfer the upper layer, ~350 μl, to a new tube.
- Add 35 μl of 3 M NaOAc and 2 μl of GlycoBlue and mix by inversion.
- Add 960 μl of 100% ethanol and mix by inversion.
- Freeze at -20 °C overnight.
SAFE STOPPING POINT: DNA should be precipitated for at least one night; this can be extended to several days without affecting recovery. - Pre-cool centrifuge to 4 °C during incubation.
- Centrifuge for 45 min/21,000 x g/4 °C and carefully discard liquid.
- Wash pellet with 1 ml 70% ice-cold ethanol.
- Spin for 10 min/21,000 x g/4 °C.
- Remove ethanol and repeat ethanol wash (steps A5j-A5k).
- Remove ethanol, spin in a microfuge and use a pipette to remove residual ethanol.
Dry at room temperature and resuspend pellet. When proceeding with LI-Capture-C, resuspend in 124 μl water. For Tag-Capture-C, the amount of water depends on the input, as several tagmentation reactions are set up in parallel (explained below). Use 20 μl water per multitude of 15,000 cells and round up (e.g., 30,000 cells:40 μl water; 50,000 cells:80 μl water).
SAFE STOPPING POINT: Store at -20 °C.
- Add 4 μl of RNase to the ligation reaction and incubate at 37 °C for 30 min.
- Quantification and QC (3 h)
Note: The NanoDrop or similar spectrophotometers are unreliable for quantifying 3C libraries due to residual DTT from the ligation buffer.- Assess 1 μl of 3C library using the Agilent Genomic ScreenTape system to determine the size distribution of library (Appendix I).
- For LI-Capture-C–take 3 μl of 3C library and 27 μl water to make a 1/10 dilution; for Tag-Capture-C–take 1 μl of 3C library and 29 μl water to make a 1/30 dilution.
Perform qPCR for digestion efficiency, using the dilution. Genomic DNA can be used as the undigested control (Appendix I). Digestion efficiency should be > 75%.
- Assess 1 μl of 3C library using the Agilent Genomic ScreenTape system to determine the size distribution of library (Appendix I).
- Fixation (2 h)
- Addition of Illumina sequencing adaptors by ligation or tagmentation to generate Capture-C libraries
LI-Capture-C
–Based on the protocol for the NEBNext Ultra DNA Library Prep Kit for Illumina (E7370)
Notes:- In this step, it is important to maintain library complexity by maximizing input DNA and minimizing DNA losses during the reactions and cleanups. When more than 1 μg of DNA is available, perform parallel preparations. These can be pooled during cleanups and at the end.
- The NEB protocol often recommends removing the DNA from the beads with a few μl more water than is necessary for the next step in the protocol. One can avoid doing this to minimize losses, but this means being very careful with the bead cleanups as contamination by the beads or ethanol can inhibit the following reaction.
- We use the Covaris S220 Focused Ultrasonicator for sonication. If using a different model sonicator, use high molecular weight gDNA to optimize sonication for a modal distribution around 200 bp in size (Appendix II).
- The Illumina indices that you put on to the library need to be complementary to the Nimblegen HE blocking oligonucleotides required in the capture step (the kits do not match; Appendix II). Buying both primer set 1 and primer set 2 eliminates waste of the more expensive Nimblegen blocking kit.
- No size selection is necessary as adaptor dimers will not be captured. All Ampure XP bead cleanups are performed with 1.8x volumes of beads.
- Sonication (1 h)
- Take 120 μl of 3C library (up to 1 μg) and transfer to a Covaris microtube. Avoid making bubbles.
- Shear DNA to 200 bp with the following settings:
Duty cycle: 10%
Intensity: 5
Cycles per burst: 200
Time: 360 sec
Set mode: Frequency sweeping - Ampure XP SPRI bead cleanup:
- Transfer reaction from the Covaris microtube to a lowBind DNA 1.5 ml microtube.
- Add 220 μl beads, pipette up and down 10 times, allow to bind at RT for 5 min.
- Place on the DynaMag, discard liquid when clear.
- Add 500 μl of fresh 80% ethanol without removing from the DynaMag. Avoid disturbing beads by running the ethanol down the front of the tube. Allow to sit on the rack for 30 sec at RT, then remove ethanol.
- Remove ethanol and add another 500 μl of fresh 80% ethanol, allow to sit for 30 sec at RT and remove all the ethanol.
- Spin down on microfuge and remove residual ethanol with a P10 pipette.
- Air dry at room temperature on the DynaMag until matt in appearance.
Note: Be careful not to over dry the beads as this will result in increased DNA losses. - Resuspend beads in 56 μl water to elute DNA, mix by pipetting 10 times.
- Incubate at RT for 2 min.
- Recover 55.5 μl for End Repair reaction.
- Transfer reaction from the Covaris microtube to a lowBind DNA 1.5 ml microtube.
- Optional: Assess 1 μl of sonicated material using the Agilent D1000 (Regular or High Sensitivity, depending on your input) ScreenTape system (Appendix II).
- Take 120 μl of 3C library (up to 1 μg) and transfer to a Covaris microtube. Avoid making bubbles.
- End repair and adaptor ligation (2 h)
- Combine 55.5 μl of DNA (5 ng-1 μg), 6.5 μl 10x End Repair Reaction Buffer (Green) and 3 μl End Prep Enzyme Mix (Green). Mix by pipetting and briefly spin down.
- Incubate in a thermocycler:
20 °C for 30 min
65 °C for 30 min - Add 15 μl Blunt/TA Ligase Master Mix (Red), 2.5 μl NEBNext Adaptor* (Red), 1 μl Ligation Enhancer (Red). Mix by pipetting and briefly spin down.
*Note: If DNA input is < 100 ng, dilute the NEBNext Adaptor for Illumina (provided at 15 μM) 10-fold in 10 mM Tris-HCl or 10 mM Tris-HCl with 10 mM NaCl to a final concentration of 1.5 μM, use immediately. - Incubate for 15 min at 20 °C in a thermocycler.
- Add 3 μl of USER Enzyme (Red). Mix by pipetting.
- Incubate for 15 min at 37 °C in a thermocycler.
- Ampure XP SPRI bead cleanup:
- Transfer reaction to a lowBind DNA 1.5 ml microtube.
- Add 160 μl beads, pipette up and down 10 times, allow to bind at RT for 5 min.
- Place on the DynaMag, discard liquid when clear.
- Add 500 μl of fresh 80% ethanol without removing from the DynaMag. Avoid disturbing beads by running the ethanol down the front of the tube. Allow to sit on the rack for 30 sec at RT, then remove ethanol.
- Remove ethanol and add another 500 μl of fresh 80% ethanol, allow to sit for 30 sec at RT and remove all the ethanol.
- Spin down in a microfuge and remove residual ethanol with a P10 pipette.
- Air dry at room temperature on the DynaMag until matt in appearance.
Note: Be careful not to over dry the beads as this will result in increased DNA losses. - Resuspend beads in 29 μl water, mix by pipetting 10 times.
- Incubate at RT for 2 min.
- Transfer 28.5 μl eluted DNA to a PCR tube.
- Transfer reaction to a lowBind DNA 1.5 ml microtube.
- Combine 55.5 μl of DNA (5 ng-1 μg), 6.5 μl 10x End Repair Reaction Buffer (Green) and 3 μl End Prep Enzyme Mix (Green). Mix by pipetting and briefly spin down.
- PCR addition of indices (2 h)
Note: Carefully choose your index to allow for multiplexing using the Illumina pooling guidelines.- Perform PCR as described below.
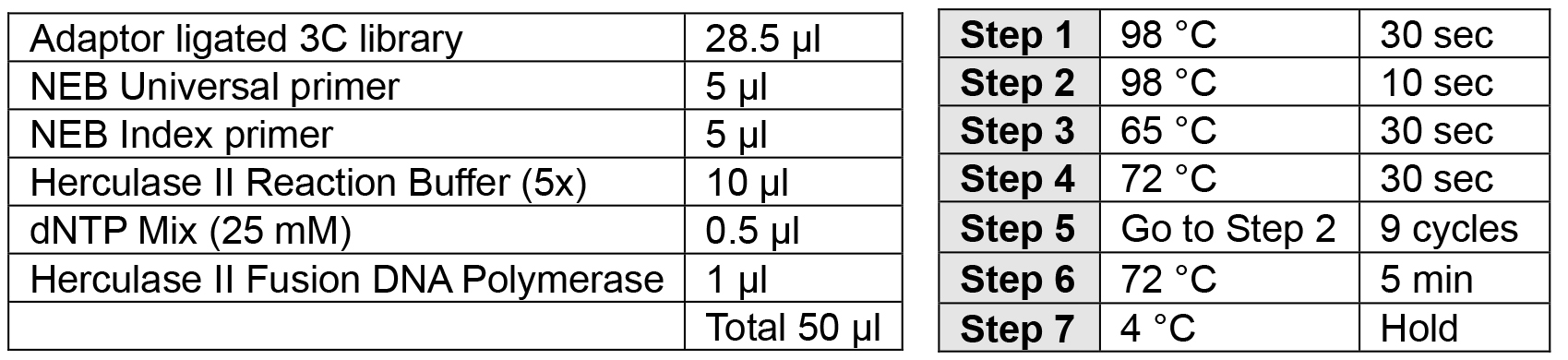
- Ampure XP SPRI bead cleanup:
- Transfer reaction to a lowBind DNA 1.5 ml microtube.
- Add 90 μl beads, pipette up and down 10 times, allow to bind at RT for 5 min.
- Place on the DynaMag, discard liquid when clear.
- Add 500 μl of fresh 80% ethanol without removing from the DynaMag. Avoid disturbing beads by running the ethanol down the front of the tube. Allow to sit on the rack for 30 sec at RT, then remove ethanol.
- Remove ethanol and add another 500 μl of fresh 80% ethanol, allow to sit for 30 sec at RT and remove all the ethanol.
- Spin down in a microfuge and remove residual ethanol with a P10 pipette.
- Air dry at room temperature on the DynaMag until matt in appearance.
Note: Be careful not to over dry the beads as this will result in increased DNA losses. - Resuspend beads in 31 μl water, mix by pipetting 10 times.
- Incubate at RT for 2 min.
- Recover 30 μl.
- Transfer reaction to a lowBind DNA 1.5 ml microtube.
- Assess 1 μl of material using the Agilent D1000 ScreenTape system (Appendix II page 3).
- Quantify library using Qubit dsDNA BR assay kit.
- Perform PCR as described below.
Tag-Capture-C
Notes:- In this step, it is important to maintain library complexity by maximizing input DNA and minimizing DNA losses during the reactions and cleanups. The maximum input for a tagmentation reaction is 50 ng. When input is higher, run several separate reactions in parallel; these can be pooled after the cleanup.
- We use custom designed index primers, for which the sequences can be found in the Appendix. We order these as TruGrade Primers from IDT.
- No size selection is necessary as adaptor dimers won’t be captured. All Ampure XP bead cleanups are performed with 1.8x volumes of beads.
- Tagmentation (1 day)
- Prepare tagmentation reactions with reagents from the Nextera DNA Sample Preparation Kit in a PCR tube:
< 50 ng 3C library in 20 μl water
25 μl TD Buffer
5 μl of TD Enzyme - Mix by pipetting, followed by a quick spin.
- Incubate at 55 °C for 12-24 h to ensure all material is tagmented.
- Zymo spin column cleanup:
- Label tubes and add 180 μl of Zymo DNA Binding Buffer.
- Add 50 μl of tagmented DNA. Gently pipette up and down 10 times to mix.
- Transfer sample mixture to a Zymo spin column in a collection tube.
- Centrifuge for 30 sec at 15,000 x g. Discard the flow-through.
- Add 200 μl Zymo DNA Wash Buffer to the column. Centrifuge for 30 sec at 15,000 x g.
- Repeat step B1d.v (Tag-Capture-C section) and centrifuge in addition 60 sec at 15,000 x g.
- Transfer the column to a 1.5 ml tube and add 34.5 μl of water to the column matrix. Incubate at RT for 2 min.
- Centrifuge for 1 min at 15,000 x g to elute the DNA.
- Label tubes and add 180 μl of Zymo DNA Binding Buffer.
- Assess 1 μl of material using the Agilent High Sensitivity D1000 ScreenTape system (Appendix III).
- Prepare tagmentation reactions with reagents from the Nextera DNA Sample Preparation Kit in a PCR tube:
- PCR addition of indices (2 h)
Note: Carefully choose your index to allow for multiplexing using the Illumina pooling guidelines.- Perform PCR as described below.
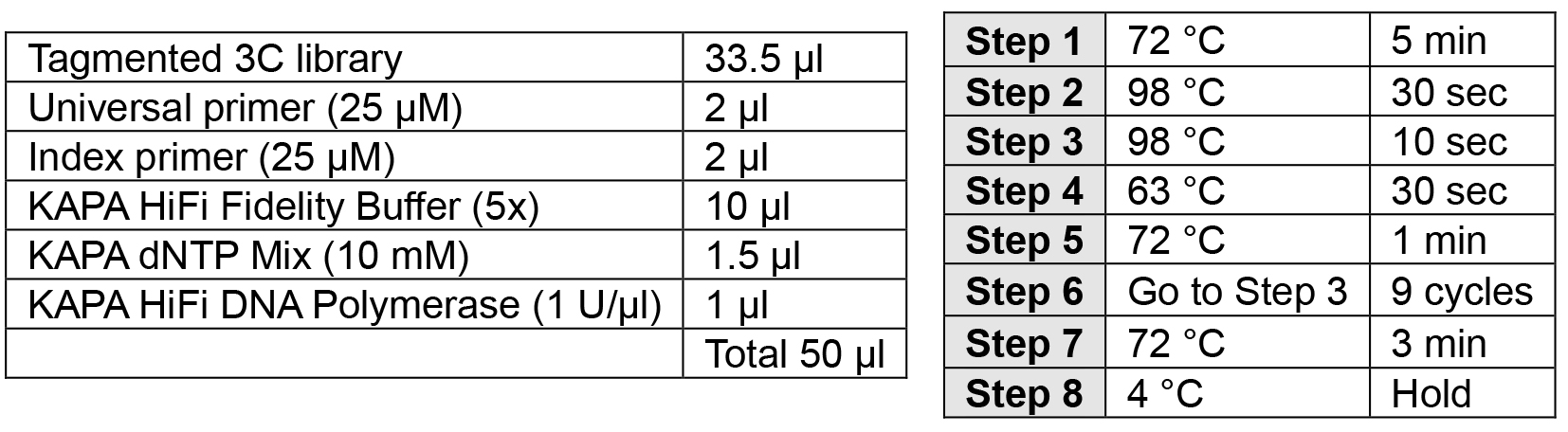
- Ampure XP SPRI bead cleanup:
- Transfer reaction to a lowBind DNA 1.5 ml microtube.
- Add 90 μl beads, pipette up and down 10 times, allow to bind at RT for 5 min.
- Place on the DynaMag, discard liquid when clear.
- Add 500 μl of fresh 80% ethanol without removing from the DynaMag. Avoid disturbing beads by running the ethanol down the front of the tube. Allow to sit on the rack for 30 sec at RT, then remove ethanol.
- Remove ethanol and add another 500 μl of fresh 80% ethanol, allow to sit for 30 sec at RT and remove all the ethanol.
- Spin down in a microfuge and remove residual ethanol with a P10 pipette.
- Air dry at room temperature on the DynaMag until matt in appearance.
Note: Be careful not to over dry the beads as this will result in increased DNA losses. - Resuspend beads in 31 μl water, mix by pipetting 10 times.
- Incubate at RT for 2 min.
- Recover 30 μl.
- Transfer reaction to a lowBind DNA 1.5 ml microtube.
- Assess 1 μl of material using the Agilent D1000 ScreenTape system (Appendix III page 4).
- Quantify library using Qubit dsDNA BR assay kit.
- Perform PCR as described below.
- In this step, it is important to maintain library complexity by maximizing input DNA and minimizing DNA losses during the reactions and cleanups. When more than 1 μg of DNA is available, perform parallel preparations. These can be pooled during cleanups and at the end.
- Enrichment for viewpoints of interest by oligonucleotide capture
–Based on the Nimblegen SeqCap SR User’s Guide Chapter 5-7
Note: The blocking oligonucleotides must be complementary to the adapters in your library. If performing LI-Capture-C, you can use the commercially available blocking oligonucleotides from Nimblegen. If you are following the Tag-Capture-C protocol, you can contact IDT and design blocking oligonucleotides using their xGen Custom Blocking Oligos service. We used a universal blocker for both the i5 adapter and i7 adapters, so we only needed 2 blocking oligonucleotides for all possible index combinations.- Hybridization preparation (1 day)
Note: Ensure capture oligonucleotides for different experiments are kept apart. The oligonucleotides are hugely in excess and tiny amounts of contamination can lead to spurious results. We would recommend that you do not order oligonucleotides that should not be mixed from the manufacturer at the same time. Avoid having buffers and blocking oligonucleotides out at the same time as the capture oligonucleotides. Oligonucleotides that are going to be pooled for a single experiment, can be ordered together. Consider ordering large designs in as a pooled set (at equimolar concentrations).
Biotinylated capture oligonucleotides- Reconstitute individual or pools of oligonucleotides to a stock concentration so that each oligo is stored at 2.9 μM (10 nmol in 3.46 μl of PCR grade water)–or any high concentration.
- Generate pools of oligonucleotides by mixing in exact 1:1 stoichiometric ratio.
Resuspend Nimblegen oligonucleotides according to the Nimblegen protocol:- Spin down tubes in a minifuge.
- Add 120 μl PCR grade water to the HE Universal Oligo tube (1 mM); vortex and spin briefly.
- Add 10 μl PCR grade water to the HE Index Oligo tube (1 mM); vortex and spin briefly.
- Add 480 μl PCR grade water to the Post-LM-PCR Oligos; vortex and spin briefly.
Resuspend the blocking oligonucleotides according to manufacturer’s instructions.
Multiplexing of samples
Mix up to 1 μg of each differentially indexed sample at exactly 1:1 ratios by mass to generate a multiplexed library. - Reconstitute individual or pools of oligonucleotides to a stock concentration so that each oligo is stored at 2.9 μM (10 nmol in 3.46 μl of PCR grade water)–or any high concentration.
- Hybridization (3 days)
- Heat vacuum centrifuge to 50 °C.
- Prepare hybridization reaction for the number of pooled libraries–up to 6 libraries are captured in a single tube. For more libraries, a master mix may be made in one tube and divided into multiple tubes.
5 μg COT DNA (5 μl of stock)/library
1 nmol TS-HE Universal Oligo (1 μl of 1 mM stock aliquot)/library
1 nmol of TS-HE Index Oligos (1 μl of 1 mM stock for each index)
up to 1 μg of each uniquely indexed 3C library
Note: For fewer than 100,000 cells you may not recover > 1 μg after indexing. Use as much as possible. - Vacuum centrifuge at 50 °C with tube lids open (rather than pierced) until sample is completely dry. Avoid drying for a long time after the liquid is gone.
- For each library in the capture reaction add:
7.5 μl 2x hybridization buffer (vial 5)
3 μl hybridization component A (vial 6) - Carefully reconstitute the DNA by pipetting and vortexing followed by briefly spinning down.
- Replace buffers and blocking oligonucleotides in freezer prior to proceeding.
- Preheat the Eppendorf ThermoMixer to 95 °C.
- Preheat a PCR block to 47 °C, the lid should be heated to 57 °C.
- Heat 4.5 μl per library of pooled biotinylated oligonucleotide probes to 47 °C in a PCR tube.
Note: The capture will be at 47 °C for ~72 h. A high-quality PCR tube is required to avoid sample loss through evaporation. - Denature the 3C library and blocking oligonucleotides by heating to 95 °C for 10 min.
- After 10 min, quickly spin the denatured library and add the entire volume to the pooled biotinylated oligonucleotides without removing from the thermocycler.
- Mix and spin briefly before replacing in the 47 °C PCR machine for 64-72 h.
- Heat vacuum centrifuge to 50 °C.
- Washing and recovery of captured material (1 day)
Notes:- Heat reagents on an Eppendorf ThermoMixer as it is more reliable than a water bath and allows the samples to be shaken.
- For capture of multiplexed libraries, scale the beads and wash buffers accordingly to the number of libraries.
- The Stringent Wash buffer, Wash Buffer I and Bead Wash Buffer are quite voluminous and may need to be made in multiple tubes. Split Wash Buffer I 1:2 as one third is heated.
- Dilute the wash buffers as per table below which allows a slight excess of each.
- Place the Stringent Wash Buffer at 47 °C on the ThermoMixer.
- Place 100 μl Wash Buffer I per captured library with an additional 20 μl at 47 °C on the ThermoMixer.
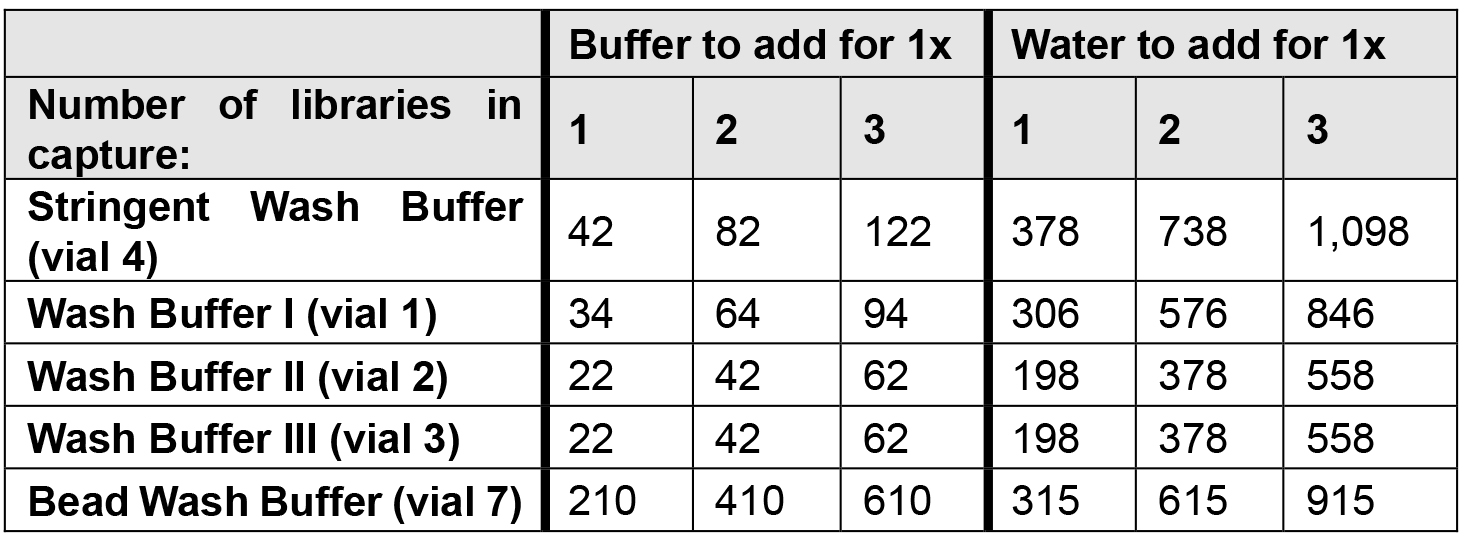
Note: The streptavidin beads will likely stick to the interior of the tube. This is particularly a problem with some makes of low bind tubes. Use tubes with minimal adhesion. We use Safeseal tubes (Sorenson) for this step.
- Place the Stringent Wash Buffer at 47 °C on the ThermoMixer.
- Prepare the streptavidin beads (M270):
- Allow the beads to heat to RT for 30 min prior to use.
- Aliquot 100 μl per captured library into a single 1.5 ml Eppendorf.
- Place on the DynaMag and remove liquid once clear.
- Add 200 μl of 1x Bead Wash Buffer per captured library and vortex to resuspend the beads, spin briefly.
- Replace on the DynaMag for 5 min and remove liquid once clear.
- Repeat wash steps (C3b.iii-C3b.v) for a total of two washes.
- Resuspend the beads in their original volume (i.e., 100 μl per captured library) with Bead Wash Buffer (1x) and aliquot into an appropriate number of tubes 1.5 ml Eppendorf tubes.
- Place on the DynaMag, remove and discard the liquid only when ready to add the capture sample from the thermocycler. Do not allow beads to dry out.
- Allow the beads to heat to RT for 30 min prior to use.
- Binding of biotinylated oligonucleotides:
- Transfer the hybridization reactions to the streptavidin beads and mix thoroughly by pipetting 10 times.
- Spin briefly if necessary to pool all of the sample in the bottom of the tube.
- Place on the ThermoMixer at 47 °C, 600 rpm for 45 min. Mix the beads with a pipette every 15 min to avoid beads settling at the bottom of the tube.
- Transfer the hybridization reactions to the streptavidin beads and mix thoroughly by pipetting 10 times.
- Washing the streptavidin beads and bound DNA:
- Add 100 μl of heated Wash Buffer I (47 °C) per captured library to the beads and bound DNA.
- Mix by vortexing.
- Place on the DynaMag, carefully discard all the supernatant when clear.
- Add 200 μl Stringent Wash buffer per captured library heated to 47 °C and mix.
- Incubate for 5 min at 47 °C.
- Place the tubes on the DynaMag, carefully discard the supernatant when clear.
- Repeat stringent wash (C3d.iv-C3d.vi) for a total of 2 washes.
- Add 200 μl Wash Buffer I (1x) at RT per captured library and mix by vortexing for 2 min. Spin briefly to ensure no sample is lost in the lid.
- Place the tubes on the DynaMag, carefully discard the supernatant when clear.
- Add 200 μl Wash Buffer II (1x) per captured library and mix by vortexing for 1 min. Spin briefly to ensure no sample is lost in the lid.
- Place the tubes on the DynaMag, carefully discard the supernatant when clear.
- Add 200 μl Wash Buffer III (1x) and mix by vortexing for 30 sec. Spin briefly to ensure no sample is lost in the lid.
- Place the tubes on the DynaMag, carefully discard the supernatant when clear.
- Remove from the DynaMag and resuspend beads in 40 μl PCR grade water per capture library. Do not discard beads–DNA is not eluted but amplified off the beads.
- Store at -20 °C or proceed to PCR amplification.
SAFE STOPPING POINT: Store at -20 °C.
- Add 100 μl of heated Wash Buffer I (47 °C) per captured library to the beads and bound DNA.
- Heat reagents on an Eppendorf ThermoMixer as it is more reliable than a water bath and allows the samples to be shaken.
- PCR amplification of captured material (1 day)
- Amplify the captured fragments in two separate reactions per captured library in a total of 14 cycles.

Note: Only use 20 μl of DNA/Bead–using 40 μl has the potential to inhibit the reaction. Two separate PCR reactions can be performed or 20 μl can be stored at -20 °C. - Ampure XP SPRI bead cleanup:
- Transfer reactions to lowBind DNA 1.5 ml microtubes. Combine up to six reactions.
- Add 90 μl beads per reaction, pipette 10 times to mix, allow to bind at RT for 5 min.
- Place on the DynaMag, discard liquid when clear.
- Add 500 μl of fresh 80% ethanol without removing from the DynaMag. Avoid disturbing beads by running the ethanol down the front of the tube. Allow to sit on the rack for 30 sec at RT, then remove ethanol.
- Remove ethanol and add another 500 μl of fresh 80% ethanol, allow to sit for 30 sec at RT and remove all the ethanol.
- Spin down in a microfuge and remove residual ethanol with a P10 pipette.
- Air dry at room temperature on the DynaMag until matt in appearance.
Note: Be careful not to over dry the beads as this will result in increased DNA losses. - Resuspend beads in 51 μl water, mix by pipetting 10 times.
- Incubate at RT for 2 min.
- Recover 50 μl.
- Transfer reactions to lowBind DNA 1.5 ml microtubes. Combine up to six reactions.
- Assess 1 μl of material using the Agilent D1000 ScreenTape system to confirm same profile as input material.
- Quantify with Qubit BR Kit.
- Amplify the captured fragments in two separate reactions per captured library in a total of 14 cycles.
- Double capture (3 days)
Note: Capture efficiency is improved by 100-1,000 fold by performing a second capture step. This increases the proportion of captured reads from 1% to 50%. This means only ~1 million reads are needed per viewpoint per sample and thus reduces sequencing requirements. Therefore, double capture is standard practice in our lab.- Pool amplified material of the parallel PCR reactions.
- Prepare the second capture hybridization reaction using all captured material (up to 2 μg) in a single reaction. Combine:
- 5 μg COT DNA (5 μl of stock)
- 1 nmol TS-HE Universal Oligo (1 μl of 1 mM stock aliquot)
- 1 nmol of TS-HE Index Oligos (1 μl of 1 mM stock in total; so if you have 4 samples, use 0.25 μl of each index for a total of 1 μl; consider making a dilution if you have many samples)
- Up to 2 μg of amplified captured material
- 5 μg COT DNA (5 μl of stock)
- Perform hybridization, washes, and amplification as described with a 24-h hybridization at 47 °C and treating the material as a single library.
- Pool amplified material of the parallel PCR reactions.
- Library quantification and sequencing (1 day)
- Use qPCR with the KAPA Library Quantification Kit to calculate the concentration of adaptor containing fragments.
Make 1:10,000 and 1:20,000 dilutions of the captured material for quantification. - Sequence using Illumina paired-end sequencing with either 150 bp or 75 bp reads (300-cycle and 150-cycle kits).
- Use qPCR with the KAPA Library Quantification Kit to calculate the concentration of adaptor containing fragments.
- Hybridization preparation (1 day)
Data analysis
The data analysis of low-input Capture-C experiments has been described (Oudelaar et al., 2017). Scripts are available in the GitHub repository (https://github.com/oudelaar/CaptureC/).
Recipes
- Fresh lysis buffer, cool to 4 °C–for 10 samples (5 ml):
10 mM Tris pH 8 (50 μl 1 M)
10 mM NaCl (10 μl of 5 M)
0.2% Igepal CA-630 (100 μl of 10%)
1x cOmplete Protease Inhibitor Cocktail (200 μl of 25x)
4.64 ml PCR grade water
Acknowledgments
The low-input Capture-C protocols are optimized adaptations from the Next-Generation Capture-C method (Davies et al., 2016). We thank the Wellcome Trust (Wellcome Trust Genomic Medicine and Statistics PhD Programme, reference 105281/Z/14/Z; Wellcome Trust Strategic Award, reference 106130/Z/14/Z) and the Medical Research Council (MRC Core Funding and Centenary Award reference 4050189188) for funding our work. The authors declare that there is no conflict of interest.
References
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. and Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10(12): 1213-1218.
- Davies, J. O., Oudelaar, A. M., Higgs, D. R. and Hughes, J. R. (2017). How best to identify chromosomal interactions: a comparison of approaches. Nat Meth 14: 125-134.
- Davies, J. O., Telenius, J. M., McGowan, S. J., Roberts, N. A., Taylor, S., Higgs, D. R. and Hughes, J. R. (2016). Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat Methods 13(1): 74-80.
- Dekker, J., Rippe, K., Dekker, M. and Kleckner, N. (2002). Capturing chromosome conformation. Science 295(5558): 1306-1311.
- Oudelaar, A. M., Davies, J. O. J., Downes, D. J., Higgs, D. R. and Hughes, J. R. (2017). Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Res 22(45): e184.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Oudelaar, A. M., Downes, D. J., Davies, J. O. and Hughes, J. R. (2017). Low-input Capture-C: A Chromosome Conformation Capture Assay to Analyze Chromatin Architecture in Small Numbers of Cells. Bio-protocol 7(23): e2645. DOI: 10.21769/BioProtoc.2645.
Category
Systems Biology > Epigenomics > Chromatin architecture
Molecular Biology > DNA > DNA structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.