- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preparation and Immunofluorescence Staining of the Trachea in Drosophila Larvae and Pupae
Published: Vol 6, Iss 9, May 5, 2016 DOI: 10.21769/BioProtoc.1797 Views: 21742
Reviewed by: Xuecai GeJihyun KimAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2014

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols



Abstract
The Drosophila melanogaster trachea is a branched network of rigid chitin-lined tubes that ramify throughout the body and functions as the fly’s respiratory organ. Small openings at the ends of the tracheal tubes allow gas exchange to occur by diffusion between internal tissues and the exterior environment. Tracheal tubes are lined by a single layer of epithelial cells, which secrete chitin and control tube morphology and size. Studies of tracheal development in Drosophila embryos have elucidated fundamental mechanisms of tube morphogenesis and maintenance in vivo, and identified major signaling pathways that regulate these processes (Manning and Krasnow, 1993; Affolter and Shilo, 2000; Zuo et al., 2013; Kerman et al., 2006; Schottenfeld et al., 2010). In recent years, there has been growing interest in the trachea during metamorphosis, when tracheal branches that had served as the respiratory organ in the larva decays and is repaired or replaced by new tracheal tissue arising from committed tracheal progenitor cells, or mature tracheal cells de-differentiated to a progenitor state (Manning and Krasnow, 1993; Sato and Kornberg, 2002; Guha et al., 2008; Guha, and Kornberg, 2005; Weaver and Krasnow, 2008; Pitsouli and Perrimon, 2010; Chen and Krasnow, 2014) forming the adult tracheal by the end of the process. The ongoing decay and tissue formation models aspects of tissue repair and regeneration in other organisms, and has been used to understand how progenitor cells divide and differentiate (Pitsouli and Perrimon, 2010; Pitsouli and Perrimon, 2013), and how they grow out of their niche to replace decaying tissue (Chen and Krasnow, 2014). Here, we present a protocol to dissect, fix, and immunostain tracheal tissue in Drosophila larvae and pupae undergoing metamorphosis. This protocol can be used to immunostain proteins expressed in tracheal tissue, or to amplify signals from weakly expressed fluorescent reporters (as shown in Figure 6). With the appropriate antibodies and genetic reporters, this protocol can be used to visualize decaying larval trachea and the progenitor cells that replace them in a time-course analysis, as well as determine expression of proteins in these cells that may play a role in tissue decay and replacement.
Materials and Reagents
- 60 mm x 15 mm petri dish [i.e., Falcon® Petri dish (Corning, catalog number: 351007 )]
- Flat-bottom 4-well dish [i.e., Nunclon® Δ Multidishes, 4 wells, flat bottom (Sigma-Aldrich, catalog number: D6789-1CS )]
- Gold SealTM Rite-OnTM Frosted microslides (VWR International, Erie Scientific, catalog number: 3050 )
- Micro cover glasses (coverslips), 22 x 22 mm Square No. 1 (VWR International, catalog number: 48366-067 )
- Black electrical tape (i.e., 3M Scotch Super 33+ Vinyl Electrical Tape 0.75 in x 450 in)
- Clear nail polish [i.e., crystal clear (Sally Hansen Hard as nails polish)]
- KimwipesTM (4.4 x 8.4 in.) (Thermo Fisher Scientific, catalog number: 06-666 )
- Austerlitz Insect Pins® ,12 mm length x 0.10 mm diameter (Minutiens in stainless steel, size 0.10 mm) (Entomoravia)
- 10 μl, 200 μl and 1,000 μl pipet tips (USA Scientific, TipOne, catalog number: 1111-3000 , 1111-0000 and 1111-2021 )
- Disposable glass Pasteur pipettes (Corning, catalog number: 7095D-5x ) with 1 ml rubber bulbs (Sigma-Aldrich, catalog number: Z111589 )
- Aluminum foil
- Parafilm M® All-Purpose laboratory film (2" x 250') (VWR International, Bemis Company, catalog number: PM992 )
- Drosophila melanogaster larvae and pupae of desired genotype raised at 25 °C
- Vials and bottles with closures [FisherbrandTM stock bottles (catalog number: AS117 ), FisherbrandTM cotton balls (catalog number: 22-456-880 ), FisherbrandTM Drosophila products, BuzzPlugsTM (catalog number: AS277 ), FisherbrandTM Drosophila vials (catalog number: AS514 )] with Drosophila food (see Cold Spring Harbor Protocols, 2014)
- Dow Corning SYLGARD® 184 Silicone Elastomer Kit [184 SIL ELAST KIT 0.5 KG (Ellsworth Adhesives)]
- 4% paraformaldehyde (PFA) diluted in PBS [i.e., 16% paraformaldehyde (VWR International, catalog number: 100503-916 ) diluted to 4% in PBS]
- Primary antibody to stain protein of interest [i.e., Chicken-anti-GFP (Abcam, catalog number: ab13970 ) to stain tracheal-expressed GFP in ppk4-Gal4, UAS-GFP larvae and pupae]
- Fluorescence conjugated secondary antibody to visualize and amplify primary antibody staining [i.e., Alexa488-conjugated Goat-anti-Chicken (Thermo Fisher Scientific, InvitrogenTM, catalog number: A-11039 ) to stain the above anti-GFP primary antibody]
- Normal serum from the same species as the secondary antibody [i.e., normal goat serum (Vector laboratories, catalog number: S-1000 )]
- Vectashield® mounting media (Vector Laboratories, catalog number: H-1000 )
- NaCl
- KCl
- Na2HPO4
- KH2PO4
- 1x Phosphate-buffered saline (PBS) (see Recipes)
- TritonTM X-100 (Sigma-Aldrich, catalog number: X100 ) diluted to 0.1% in PBS (see Recipes)
- Block solution (see Recipes)
- DAPI staining solution (see Recipes)
- Dissection dish (see Recipes)
Equipment
- Incubator to house Drosophila set to 25 °C
- Stereomicroscope with light source [i.e., Carl ZeissTM StemiTM 2000C with KL 300 LED Cold Light Source (120 V) (Thermo Fisher Scientific, catalog number: 12-070-284 )]
- Dumont #5 mirror finish forceps biology tips/straight/inox/11 cm (Fine Science Tools, catalog number: 11252-23 )
- Vannas spring scissors-straight/sharp/8 cm/3 mm cutting edge (Fine Science Tools, catalog number: 15000-00 )
- Benchtop shaker (i.e., Bellco Glass 7744-06115 Mini Orbital Shaker)
- Clay AdamsTM Nutator Mixer (BD, catalog number: 421105 )
- P2, P20, P100, and P1000 Pipetman® Pipettes (Gilson Scientific Ltd., catalog number: F144801 , F123600 , F123615 and F123602 )
- Small watercolor paintbrush, round, size 0
- Stainless steel spatula with a micro spoon end (Ted Pella Inc., catalog number: 13500 )
Procedure
- Dissection of larvae and pupae by ventral filleting
- Pick larvae and pupae of the appropriate age. Tracheal progenitors are activated during the late second-instar (L2) stage and remodel the trachea throughout the third-instar (L3) and pupal stages.
- The staging of Drosophila melanogaster larvae and pupae is described in (Ashburner, 2005).
- Young (L1 to early L3 stage) larvae [see Ashburner (2005) for more information on larval staging] will burrow into the food as they feed. To isolate these larvae, scoop out a portion of the food with the spoon end of the spatula and place it into a petri dish filled with PBS. Gently separate the food with the spatula until larvae can be seen floating in the PBS, and then transfer the larvae into a new PBS-filled petri dish with a small paintbrush or a glass Pasteur pipette.
- Late L3 stage larvae that are about to enter puparium formation will migrate onto the walls of the vial or bottle they are cultured in, and because of this behavior are commonly referred to as wandering L3 larvae. Wet a small paintbrush by dipping briefly in PBS and blot the excess PBS on a KimwipeTM. Gently pick up larvae from the wall of the vessel with the tip of the brush. Dip the tip of the brush with the larvae into a petri dish filled with PBS and move the brush back and forth to allow the larvae to be transferred to the PBS.
- During the pupal stages, tracheal metamorphosis occurs rapidly, and so it is necessary to obtain pupae of precise age. To do this, locate animals that have just entered pupariation [0 h after puparium formation (APF)]; they should be white, attached to the side of the vessel, and immobile (Ashburner, 2005) (Figure 1A). Gently detach these pupae with a small wet paintbrush and transfer them into a new vial. Set the pupae onto the walls of the vial so that the ventral surface of the animal is attached to plastic (the way they normally attach to the vessel wall). Incubate at 25 °C until the desired time APF has been reached, then gently detach and pick up the pupae with a small wet paintbrush and transfer to a dissection dish. When transferring, be careful not to damage the pupae by putting too much pressure on them.
- The staging of Drosophila melanogaster larvae and pupae is described in (Ashburner, 2005).
- Preparations for dissection.
- Using forceps pick up insect pins one at a time and insert them vertically tip down into the silicone elastomer layer of the dissection dish to be used. Insert just deep enough that the pins are held in place by the silicone elastomer. This way, pins can be easily retrieved with forceps and moved to where they are needed during dissection.
- Ventral-filleting larvae, and pupae younger than 12 h APF (before eversion of the head) (see Videos 1 and 2).
 Video 1. Dissecting larva Video 2. Dissecting pupa younger than 12 h APF
Video 1. Dissecting larva Video 2. Dissecting pupa younger than 12 h APF- Place Petri dishes containing larvae on ice for 5 to 10 min. The cold temperature will slow the larva’s movements and facilitate dissection. Do not leave larvae on ice for more than 45 min. Pupae are immobile and will not need to be exposed to cold temperature prior to dissection.
- Fill a dissection dish with cold PBS and move the larva or pupa into the dissection dish.
- With forceps, turn the animal ventral side up, and gently hold it in place using forceps. Locate the posterior (Figure 2A and 3A, white arrows) of the left and right tracheal dorsal trunks, a set of large air-filled tubes running along the anterior-posterior axis on the dorsal side of the animal, and insert a pin between the trunks at the posterior (Figure 2A and 3A, white arrowheads) into the silicone elastomer. Insert another pin (Figure 2A and 3A, black arrowheads) between the dorsal trunks at the anterior end of the animal (Figure 2A and 3A, black arrows). When dissecting larvae, first stretch the animal by pulling on the anterior with forceps, and then insert the second pin just below the pigmented mouthparts.
- Gently pull up on the ventral epidermis (and cuticle in pupae) with forceps, and carefully insert the bottom blade of the Vannas spring scissors where the posterior pin has been inserted, and slide the blade just under the ventral epidermis. If necessary, enlarge the opening made by the pin by pulling on the epidermis with forceps before inserting the scissor blade. Cut towards the anterior of the animal (Figure 2A and 3A, horizontal dashed lines). Pupae can also be cut from anterior to posterior, although larvae are more easily cut from posterior to anterior.
- Grasp the edges of the cut epidermis (and cuticle in pupae) with forceps, pull gently to the side and secure with pins (Figure 2B-C and 3B-C; asterisks) so that the epidermis is one flat layer (Figure 2C and 3C). Make additional cuts to the epidermis and cuticle as necessary (Figure 3A vertical dashed lines) to allow the sample to be pinned flat. Be careful to not pin or damage tracheal branches during the process.
- Remove internal organs by pulling on them with forceps. Cut any terminal tracheal branches attached to the tissue (Figure 2D, arrows) so that the tissue can be removed.
- The main tracheal branches [as described in Manning and Krasnow (1993)] should now be visible (Figure 2C, E and 3C); they are rigid air-filled tubes and attached to the epidermis. Clear remaining debris and tissue around tracheal branches by gently pipetting PBS onto the sample with a Pasteur pipette.
- Place Petri dishes containing larvae on ice for 5 to 10 min. The cold temperature will slow the larva’s movements and facilitate dissection. Do not leave larvae on ice for more than 45 min. Pupae are immobile and will not need to be exposed to cold temperature prior to dissection.
- Ventral-filleting pupae older than 12 hours APF (after head eversion) (see Video 3). Video 3. Dissecting pupa older than 12 h APF
- Pupae that have completed the larval-pupal molt and everted their heads (Figure 1C), which occurs around 12 h APF, need to be removed from the pupal case prior to dissection. To do this, move the pupa onto a dry dissection dish ventral side down. Work under a stereomicroscope, and hold a pair of Dumont #5 forceps in each hand. Carefully hold the pupa with forceps using the non-dominant hand and use forceps in the dominant hand to insert the tip of an insect pin through the posterior end of the pupal case just below where the pupa has detached from the pupal case (Figure 4A, white arrowhead). Continue inserting the pin into the silicone elastomer layer of the dish; the pupa should be securely pinned to the dish. Using Vannas spring scissors make a horizontal cut through both layers of the pupal case at the anterior end above the pupa’s head (Figure 4A, vertical dashed line). Remove the rest of the pupal operculum with forceps. Gently insert scissors just under the pupal case along the anterior-posterior axis (careful not to damage the pupa inside), and cut the pupal case from anterior to the posterior (Figure 4A, horizontal dashed line). Carefully peel away the pupal case and remove it or pin it down to the dish (Figure 4C, asterisks), making extra cuts in the pupal case as necessary to relieve any pressure exerted by the case on the pupa inside. Gently pick up the pupa from the pinned case with a small wet brush and transfer to a new dissection dish. An alternative method to remove the pupa from the pupal case is described in Wang and Yoder (2011).
- With a wet brush (or very gently with forceps) orient the pupa ventral side up. Gently holding the pupa in place with forceps, insert a pin through the head (Figure 4D, black arrowhead).
- With forceps, pull up on the ventral epidermis at the opening made by the pin and slide the bottom blade of the Vannas spring scissors just under the ventral epidermis. Cut all the way to the posterior of the animal (Figure 4D, black dashed line).
- Make a small cut in the dorsal epidermis from posterior to anterior just to the midpoint of the abdomen (Figure 4D, black dotted line). This cut allows the pupa to be pinned flat.
- Grasp the cut edges of the ventral epidermis with forceps and pin down so that the epidermis is one flat layer (Figure 4E).
- Much of the internal tissues of the pupa will be histolyzed and can be removed by gently pipetting PBS onto the sample with a Pasteur pipette. Pipetting in the anterior to posterior direction results in least amount of damage to the tracheal branches. Any remaining tissue can be gently pulled out with forceps.
- Pupae that have completed the larval-pupal molt and everted their heads (Figure 1C), which occurs around 12 h APF, need to be removed from the pupal case prior to dissection. To do this, move the pupa onto a dry dissection dish ventral side down. Work under a stereomicroscope, and hold a pair of Dumont #5 forceps in each hand. Carefully hold the pupa with forceps using the non-dominant hand and use forceps in the dominant hand to insert the tip of an insect pin through the posterior end of the pupal case just below where the pupa has detached from the pupal case (Figure 4A, white arrowhead). Continue inserting the pin into the silicone elastomer layer of the dish; the pupa should be securely pinned to the dish. Using Vannas spring scissors make a horizontal cut through both layers of the pupal case at the anterior end above the pupa’s head (Figure 4A, vertical dashed line). Remove the rest of the pupal operculum with forceps. Gently insert scissors just under the pupal case along the anterior-posterior axis (careful not to damage the pupa inside), and cut the pupal case from anterior to the posterior (Figure 4A, horizontal dashed line). Carefully peel away the pupal case and remove it or pin it down to the dish (Figure 4C, asterisks), making extra cuts in the pupal case as necessary to relieve any pressure exerted by the case on the pupa inside. Gently pick up the pupa from the pinned case with a small wet brush and transfer to a new dissection dish. An alternative method to remove the pupa from the pupal case is described in Wang and Yoder (2011).
- Fix the dissected larvae and pupae.
- Remove the PBS containing detached internal tissues with a P1000 pipet. Gently pipet 1 to 2 ml of 4% PFA along the sides of the dish, enough to cover all of the samples.
- Place the dissection dish on an orbital shaker in the fume hood. Cover samples with a light protected box if samples express fluorescent proteins. Set the shaker on a medium-low setting (i.e., 3 or 4) and allow the PFA to fix the samples at room temperature for 30 min.
- Remove the PBS containing detached internal tissues with a P1000 pipet. Gently pipet 1 to 2 ml of 4% PFA along the sides of the dish, enough to cover all of the samples.
- Remove the 4% PFA and properly dispose as hazardous waste. Gently pipet 1 to 2 ml room temperature PBS, and shake for 5 minutes to wash out residual PFA. Repeat 2 more times.
- With forceps and working under the stereomicroscope, carefully remove the pins holding the samples in place. Transfer the fixed samples into a 4-well dish for staining. Up to 6 larvae and 8 pupae can be stained in each well.
- Proceed with the staining procedure described below or store the samples immersed in PBS at 4 °C with the plate wrapped in Parafilm. Samples can be stored for up to a few weeks.
- Pick larvae and pupae of the appropriate age. Tracheal progenitors are activated during the late second-instar (L2) stage and remodel the trachea throughout the third-instar (L3) and pupal stages.
- Immunofluorescence staining larval and pupal trachea
- Permeabilize the fixed sample(s) by incubating in 650 μl PBS with 0.1% TritonTM X-100 at room temperature for 5 min. Pipet in liquids gently along the sides of each well to prevent damaging the samples. If samples express a fluorescent reporter, cover the dish with aluminum foil or a light protected box for this and subsequent steps.
- Remove the PBS with 0.1% TritonTM X-100, add 650 μl block solution into each well, and incubate at room temperature for 30 min on a Nutator mixer.
- Dilute primary antibody in block solution. Remove the block solution from the step above and apply 650 μl of the primary antibody solution per well. If amount of the antibody solution needs to be conserved, add just enough to immerse the samples, about 250 μl per well. Incubate overnight at 4 °C on a Nutator mixer.
- Remove the antibody solution, add 650 μl PBS 0.1% Triton, and incubate on a Nutator mixer for 5 min at room temperature to wash away residual antibodies. Remove wash solution, and repeat 2 more times.
- Wash the samples 3 more times in 650 μl PBS 0.1% Triton for 30 min each. These longer washing steps are necessary to remove any antibodies that might have been trapped in the tracheal tubes.
- Dilute fluorescence conjugated secondary antibody in PBS 0.1% Triton, and apply 250 to 650 μl to each well. Incubate on a Nutator mixer for 1 h at room temperature. Cover the dish with aluminum foil or a light protective box for this and subsequent steps.
- Wash the samples in PBS 0.1% Triton 3 times for 5 min each.
- Apply 650 μl DAPI staining solution each well and incubate for 10 to 20 min.
- Remove the DAPI solution, and wash the samples with PBS 0.1% Triton 3 times for 5 min each, and then 3 times for 30 min each.
- Permeabilize the fixed sample(s) by incubating in 650 μl PBS with 0.1% TritonTM X-100 at room temperature for 5 min. Pipet in liquids gently along the sides of each well to prevent damaging the samples. If samples express a fluorescent reporter, cover the dish with aluminum foil or a light protected box for this and subsequent steps.
- Mount samples on glass slides
- Cut two pieces of black electrical tape about 20 mm x 5 mm and affix to the glass slide. Leave a space slightly less than the width of the coverslip between the two pieces of tape (Figure 5A). The pieces of tape are used to create a small space between the slide and the coverslip (Figure 5B); if the samples are mounted without the tape, the coverslip may crush the sample and damage tracheal structures.
- Add two to three drops of Vectashield® mounting media onto the glass slide between the two pieces of tape. Place your samples in the Vectashield® and adjust the orientation of the samples so that the internal surface of the sample containing the tracheal branches is on top and the exterior surface of the epidermis is on the bottom. To facilitate imaging, orient the samples on the slide so that when observing the slide through the microscope the anterior is on the left and the posterior on the right. With forceps, remove any dust particles that might be lodged in the sample.
- Cover samples with a coverslip and allow the Vectashield® to fill the space between the slide and coverslip, gently push down on the coverslip with forceps if necessary. Remove any extra Vectashield leaking from under the coverslip with a Kimwipe.
- Apply clear nail polish around the edges of the coverslip to seal in the samples and Vectashield®. Cover the slides with a light protective box, and allow the nail polish to dry at room temperature (approximately 10 min).
- Proceed with microscopic analysis, or store the slides at 4 °C.
- Cut two pieces of black electrical tape about 20 mm x 5 mm and affix to the glass slide. Leave a space slightly less than the width of the coverslip between the two pieces of tape (Figure 5A). The pieces of tape are used to create a small space between the slide and the coverslip (Figure 5B); if the samples are mounted without the tape, the coverslip may crush the sample and damage tracheal structures.
Representative data

Figure 1. Appearance of Drosophila melanogaster pupae. A. Pupae that have just entered puparium formation (0 h APF) are white, immobile, and attached to the sides of the culture vessel. B. As the pupa ages, the cuticle tans and turns from white to a yellow brown color. C. Pupae undergoing the larval-pupal molt detach from the pupal case. The molt is completed by head eversion (around 12 h APF), and a gap can be seen between the pupal case and the head of the pupa inside (white arrow). Scale bar, 500 µm
Figure 2. Dissecting Drosophila melanogaster larvae by ventral filleting. A. Larvae are first pinned ventral side up between the tracheal dorsal tracheal trunks at the posterior end (white arrows; position of pin indicated by the white arrowhead). A second pin is inserted just under the pharynx at the anterior end (black arrowhead) between the left and right anterior dorsal trunks (black arrows). The ventral epidermis is then cut along the anterior-posterior axis (black dashed line). B. The cut epidermis is pulled to the side and pinned down to the dissection dish (asterisk). C. Internal tissues are pulled out by forceps; thin terminal branches of the trachea attached to the tissues are cut (arrows) to allow the tissue to be removed. D. As internal tissues are removed, more pins (asterisks) are inserted to pin the epidermis flat. The major branches of the trachea are visible after removal of the internal tissues (enlarged in E). DB, dorsal branch; DT, dorsal trunk; TC; transverse connective; LT, lateral trunk. Tracheal progenitor cells (arrow) in the fourth (Tr4) and fifth (Tr5) tracheal metameres are often visible in wandering L3 larvae, appearing as a small patch of tissue located at the junction of the TC with the DT. Scale bars, 500 µm (A-C), 250 µm (D-E) 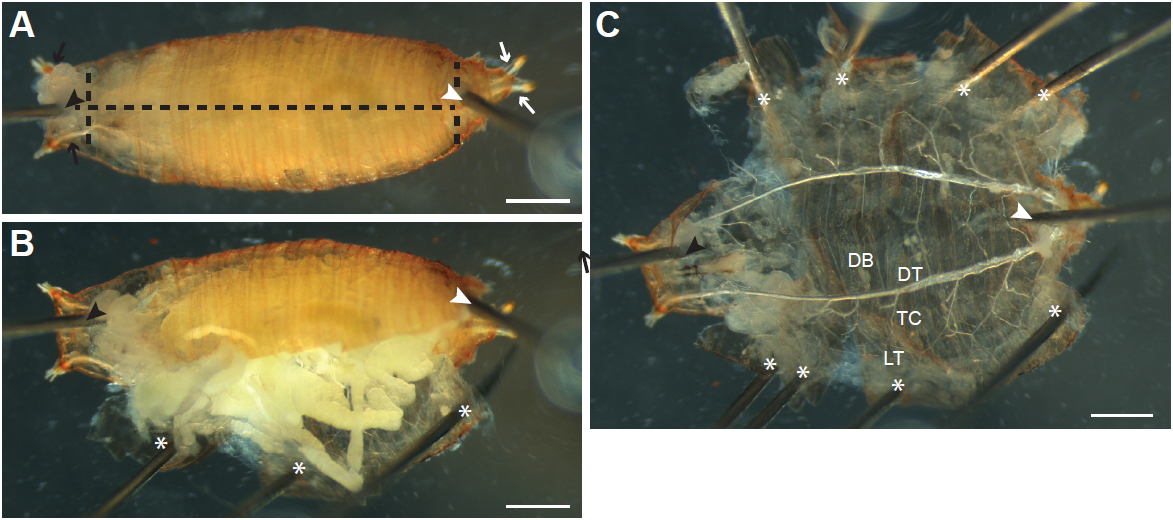
Figure 3. Dissecting young pupae (younger than 12 h APF). A. Young pupae that have not yet completed the larval-pupal molt and everted their heads (0 to 12 h APF) are dissected by pinning at the anterior (black arrowhead) and posterior (white arrowhead) at a position between the left and right dorsal trunks (arrows). Cuts are then made through the pupal case and ventral epidermis (black dashed lines). B and C. Additional pins (asterisks) are inserted to pin the sample flat. Internal organs and tissues are removed by pulling with forceps and small cuts made by spring scissors. C. After removal of internal tissue, tracheal branches are visible. DB, dorsal branch; DT, dorsal trunk; TC; transverse connective; LT, lateral trunk. Scale bars, 500 µm 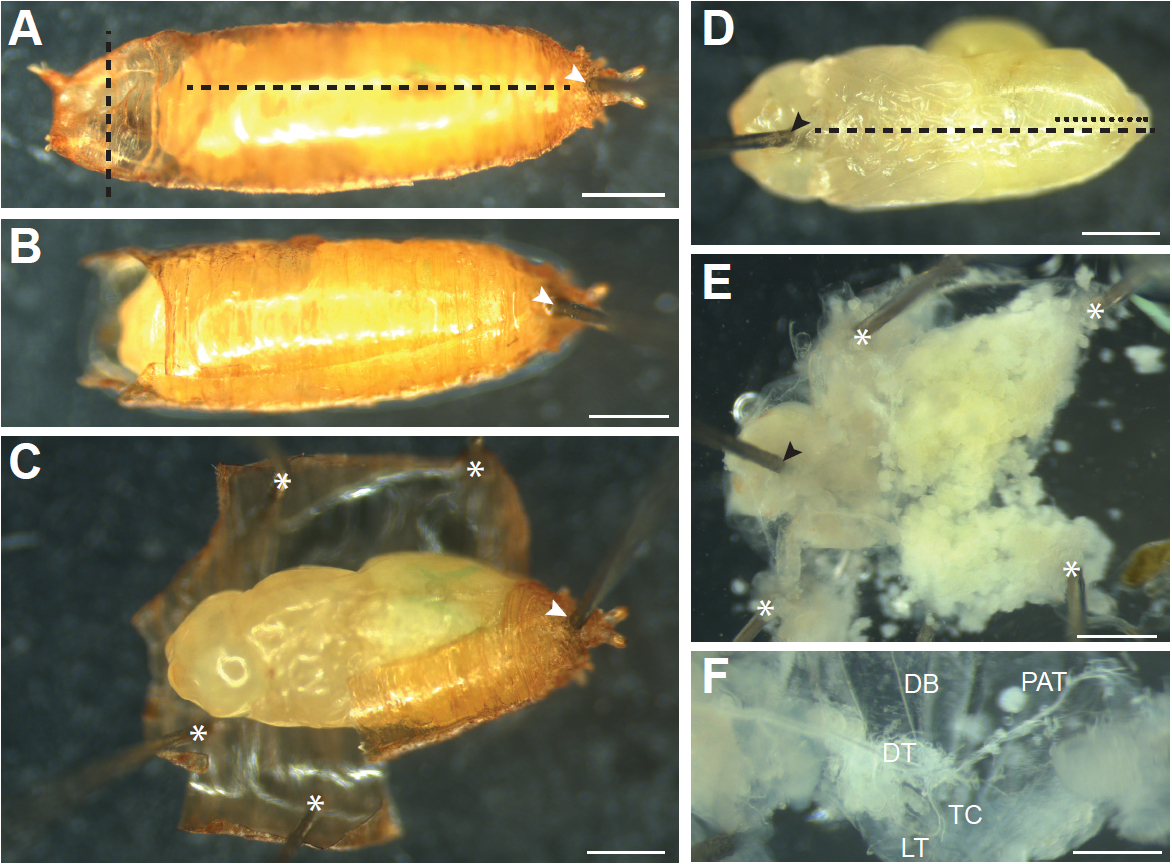
Figure 4. Dissecting older pupae (older than 12 h APF). A. Pupae that have everted their heads need to be removed from the pupal case. First a pin is inserted through the posterior end of the pupal case in the region where the pupa has detached from the case (white arrowhead) to secure the pupa to the dissection dish. Cuts are then made through the pupal case just above the head (vertical black dashed line) and through the dorsal pupal case from anterior to posterior (horizontal black dashed line). B. A pupal after the cuts through pupal case have been made. C. The pupal case is then peeled away and held down with pins (asterisks) until the pupa can be picked up with a brush and transferred to a new dissection dish. D. The pupa is oriented ventral side up, and a pin is inserted into the head (black arrowhead). Cuts through the ventral (black dashed line) and dorsal (black dotted line) epidermis are made and the epidermis is pinned down until it is flat, as shown in (E). Additional pins used to pin the sample down are indicated by asterisks. Internal tissues are removed by forceps or gentle pipetting of PBS onto the sample to expose the trachea. F. Main branches of the pupal trachea. DB, dorsal branch; DT, dorsal trunk; TC, transverse connective; LT, lateral trunk; PAT, pupal abdominal trachea. Scale bars, 500 µm (A-E), 250 µm (F) 
Figure 5. Mounting samples on glass slides. A. To prevent the coverslip from crushing the tracheal in larval and pupal samples, two small pieces of electrical tape (arrows) are affixed to the slide. B. The tape props the coverslip up, creating a small space between the slide and coverslip. 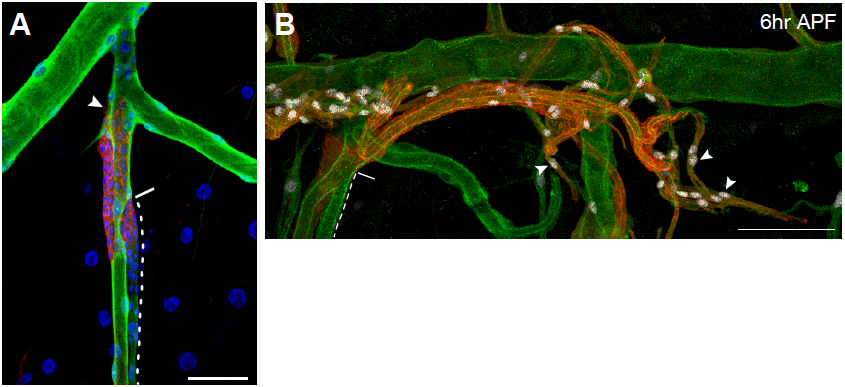
Figure 6. Representative images of immunostained larvae and pupae. A. Tracheal metamere 5 (Tr5) of a third-instar ppk4-Gal4, UAS-GFP; btl-RFP-moe larva in which larval tracheal cells have been immunostained for the GFP reporter expressed in these cells (green), and outgrowing progenitor cells (arrowhead) are immunostained for the membrane-associated RFP reporter they express (red). Nuclei are stained with DAPI (blue). Dashed line outlines the progenitor niche; solid line indicates the niche exit. B. Tr5 and Tr6 in a btl-RFP-moe/ crumbs::GFP pupa 6 h after puparium formation. Signals from the RFP reporter expressed in progenitor cells and GFP expressed on the apical surface of trachea are amplified by immunostaining for RFP (red) and GFP (green), respectively. Expression of the Pruned SRF protein in trachea progenitors (arrowheads) is detected by an anti-Pruned antibody (white). Scale bar, 50 µm (A), and 100 µm (B). Image taken from a previously published study (Chen and Krasnow, 2014).
Notes
- Young larvae (first and young second instar larvae) may be too small to be dissected by the method described in this protocol.
- Tracheal tubes can sometimes trap antibodies, leading to false positive staining. Try to include negative controls in experiments, such as samples incubated with secondary antibody but not primary antibody, or samples that do not express the epitope detected by the primary antibody.
- Steps B3 to B5 can be skipped if using a primary antibody directly conjugated to a fluorophore.
- Staining of the tracheal lumen with a fluorescence-labeled chitin binding probe or wheat germ agglutinin (Weaver and Krasnow, 2008; Chen and Krasnow, 2014) can be accomplished with this protocol, skipping steps B3 to B5.
- The lumens of tracheal branches autofluoresce when observed through the UV channel. This autofluorescence can be used to determine outlines of the tracheal branches, and is distinguishable from nuclear DAPI stains. However, to avoid confusing antibody stains with the autofluorescence, secondary antibodies conjugated to fluorescent reporters that fluoresce at other wavelengths should be used.
- Alternative detergent solutions (i.e., 0.1% Tween-20 diluted in PBS) can be used to permeabilize cell membranes in the samples. Permeabilizing samples with 0.1% Triton X-100 was chosen because it was optimal for staining both the tracheal lumen and tracheal cells. 0.1% Tween-20 resulted in good staining in tracheal cells but weak staining in tracheal lumens.
Recipes
- Recipe for 1x phosphate-buffered saline (PBS)
NaCl 8 g
KCl 0.2 g
Na2HPO4 1.44 g
KH2PO4 0.24 g
Dissolve in 800 ml H2O, adjust pH to 7.4, add H2O to 1 L final volume. - 0.1% TritonTM X-100 PBS solution
Extract 5 ml TritonTM X-100 with a 6 ml syringe and mix into 45 ml of PBS
Vortex until completely dissolved to make 10% TritonTM X-100 stock solution
Dilute 2.5 ml 10% stock solution in PBS (250 ml final volume) to make 0.1% solution - Block solution
0.1% TritonTM X-100 PBS solution
10% normal serum from the species of the secondary antibody (i.e., normal goat serum) - DAPI staining solution
0.1 µg/ml DAPI diluted in PBS - Dissection dish
Mix components of the SYLGARD® 184 Silicone Elastomer Kit following the manufacturer's instructions
Apply enough of the mixture to the bottom of a 60 mm x 15 mm Petri dish to cover the bottom of the dish with a 1 cm thick layer
Let the mixture cure overnight at room temperature before use
Acknowledgments
This protocol is adapted from methods developed by others and used in previous studies (Weaver and Krasnow, 2008; Pitsouli and Perrimon, 2010; Pitsouli and Perrimon, 2013; Levi et al., 2006). Funding was provided by the Howard Hughes Medical Institute (HHMI), a Genentech Graduate Fellowship, and a Ruth L. Kirschstein NIH training grant.
References
- Affolter, M. and Shilo, B. Z. (2000). Genetic control of branching morphogenesis during Drosophila tracheal development. Curr Opin Cell Biol 12(6): 731-735.
- Ashburner, M., Golic, K. G.and Hawley, R. S. (2005). Chapter 6: Life Cycle. In: Ashburner, M. (ed). Drosophila: a laboratory handbook. Cold Spring Harbor Laboratory Press, pp 150-158.
- Chen, F. and Krasnow, M. A. (2014). Progenitor outgrowth from the niche in Drosophila trachea is guided by FGF from decaying branches. Science 343(6167): 186-189.
- Cold Spring Harbor Protocols (2014). Fly Food.
- Guha, A. and Kornberg, T. B. (2005). Tracheal branch repopulation precedes induction of the Drosophila dorsal air sac primordium. Dev Biol 287(1): 192-200.
- Guha, A., Lin, L. and Kornberg, T. B. (2008). Organ renewal and cell divisions by differentiated cells in Drosophila. Proc Natl Acad Sci U S A 105(31): 10832-10836.
- Kerman, B. E., Cheshire, A. M. and Andrew, D. J. (2006). From fate to function: the Drosophila trachea and salivary gland as models for tubulogenesis. Differentiation 74(7): 326-348.
- Levi, B. P., Ghabrial, A. S. and Krasnow, M. A. (2006). Drosophila talin and integrin genes are required for maintenance of tracheal terminal branches and luminal organization. Development 133(12): 2383-2393.
- Manning, G. and Krasnow, M. A. (1993). In: Martinez-Arias, A. and Bate, M. (eds). The Development of Drosophila. Cold Spring Harbor Laboratory Press, 609-685.
- Sato, M. and Kornberg, T. B. (2002). FGF is an essential mitogen and chemoattractant for the air sacs of the Drosophila tracheal system. Developmental cell 3(2): 195-207.
- Schottenfeld, J., Song, Y. and Ghabrial, A. S. (2010). Tube continued: morphogenesis of the Drosophila tracheal system. Curr Opin Cell Biol 22(5): 633-639.
- Pitsouli, C. and Perrimon, N. (2010). Embryonic multipotent progenitors remodel the Drosophila airways during metamorphosis. Development 137(21): 3615-3624.
- Pitsouli, C. and Perrimon, N. (2013). The homeobox transcription factor cut coordinates patterning and growth during Drosophila airway remodeling. Sci Signal 6(263): ra12.
- Wang, W. and Yoder, J. H. (2011). Drosophila pupal abdomen immunohistochemistry. J Vis Exp 56: e3139.
- Weaver, M. and Krasnow, M. A. (2008). Dual origin of tissue-specific progenitor cells in Drosophila tracheal remodeling. Science 321(5895): 1496-1499.
- Zuo, L., Iordanou, E., Chandran, R. R. and Jiang, L. (2013). Novel mechanisms of tube-size regulation revealed by the Drosophila trachea. Cell Tissue Res 354(2): 343-354.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chen, F. (2016). Preparation and Immunofluorescence Staining of the Trachea in Drosophila Larvae and Pupae. Bio-protocol 6(9): e1797. DOI: 10.21769/BioProtoc.1797.
Category
Developmental Biology > Morphogenesis > Metamorphosis
Cell Biology > Tissue analysis > Tissue staining
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.